Computational Biology
BIOSC 1540
Fall 2024 • University of Pittsburgh • Department of Biological Sciences
This course offers a systematic examination of foundational concepts and an in-depth understanding of the significant fields of computational biology. Students learn about genomics, transcriptomics, computer-aided drug design, and molecular simulations in the context of current research applications. This course is the first stepping stone for undergraduates at the University of Pittsburgh before taking Computational Genomics (BIOSC 1542) or Simulation and Modeling (BIOSC 1544).
Contributions¶
All comments, questions, concerns, feedback, or suggestions are welcome!
License¶
Code contained in this project is released under the GPLv3 license as specified in LICENSE.md.
All other data, information, documentation, and associated content provided within this project are released under the CC BY-NC-SA 4.0 license as specified in LICENSE_INFO.md.
Why are we using copyleft licenses? Many people have dedicated countless hours to producing high-quality materials incorporated into this website. We want to ensure that students maintain access to these materials.
Web analytics¶
Why would we want to track website traffic?
An instructor can gain insights into how students engage with online teaching materials by analyzing web analytics. This information is instrumental in assessing the effectiveness of the materials. Web analytics reveal the popularity of specific topics or sections among students and empower instructors to tailor future lectures or discussions. Analytics also provides valuable data for curriculum development, helping instructors identify trends, strengths, and weaknesses in course materials. Additionally, instructors may leverage web analytics as evidence of their commitment to continuous improvement in teaching methods, which is helpful in discussions related to professional development, promotions, or tenure.
We track website traffic using plausible, which is privacy-friendly, uses no cookies, and is compliant with GDPR, CCPA, and PECR.
Syllabus ↵
Syllabus¶
Semester: Fall 2024
Meeting time: Tuesdays and Thursdays from 4:00 - 5:15 pm.
Location: 1501 Posvar
Instructor: Alex Maldonado, PhD (he/him/his)
Email: alex.maldonado@pitt.edu
Office hours:
| Day | Time | Location |
|---|---|---|
| Tuesday | 11:30 am - 12:30 pm | 102 Clapp Hall |
| Thursday | 11:30 am - 12:30 pm | 315 Clapp Hall |
Catalog description¶
This course gives students a broad understanding of how computational approaches can solve problems in biology. We will also explore the biological and computational underpinnings of the methods.
Note
The catalog course description mandates what material this course has to cover. How the material is covered is at the discretion of the instructor.
Prerequisites¶
To enroll, you must have received a minimum grade of C in Foundations of Biology 2 (BIOSC 0160, 0165, or 0716).
Programming experience
In contrast to previous semesters, no programming will be necessary for the successful completion of this course.
Course philosophy¶
The Computational Biology course explores how to gain insight into biological phenomena with computational methodologies. My teaching philosophy that guides this course is below.
Critical thinking is paramount¶
Critical thinking and problem-solving are essential in education, especially in computational biology. My teaching approach is centered around engaging students in real-world scenarios and challenges. This requires them to apply, analyze, and synthesize information and helps them understand the practical application of computational biology, moving away from rote memorization. This approach fosters a deep understanding of the subject matter, encouraging students to explore the complexities and interconnectedness of biological systems through computational methods. By encouraging inquiry, debate, and collaboration, I aim to equip students with the skills necessary to navigate and contribute to the ever-evolving landscape of computational biology. The goal is not just to impart knowledge but to cultivate innovative thinkers and problem solvers who are prepared to tackle the challenges of the future.
Unlike traditional biology courses such as foundations of biology or biochemistry, this computational biology class stands out with its unique learning approach, resembling a computer science course. While the problems and concepts explored may have roots in similar biological foundations, the course's computational nature demands a distinct set of skills and mindsets. As part of this course, students will be expected to take the initiative in their learning and problem-solving. They will encounter open-ended challenges that transcend the boundaries of a single discipline, fostering their ability to think critically and independently. This course aims to foster a spirit of independent inquiry and adaptability, preparing students to thrive in the multidisciplinary and rapidly evolving field of computational biology.
Info
In traditional biology courses, such as foundations of biology or biochemistry, students often learn through rote memorization of facts, concepts, and processes. For example, students might be asked to memorize the steps of DNA replication or the names and functions of various enzymes involved in cellular metabolism. While this knowledge is essential, it only sometimes challenges students to apply their understanding to novel situations or develop problem-solving skills.
In contrast, a computational biology course focuses on problem-based and open-ended learning, which is more akin to the approach in computer science courses. Students are presented with complex, real-world biological problems that require them to apply their knowledge innovatively and develop their own computational solutions. For instance, students might be asked to analyze large genomic datasets to identify patterns or anomalies, develop algorithms to predict protein structures, or create models to simulate the spread of infectious diseases.
These open-ended problems necessitate a deep understanding of biological concepts and the ability to translate that knowledge into computational frameworks. Students must think critically, break complex problems into manageable components, and develop creative solutions. They often need to engage in self-driven learning, exploring new computational tools and techniques to tackle the challenges.
A computational biology course emphasizes problem-based and open-ended learning, cultivating essential critical thinking, creativity, and adaptability skills. These skills prepare students for careers in computational biology and equip them with the tools to tackle the novel challenges they will face in any scientific field.
Learning happens outside your comfort zone¶
This computational biology course embraces the idea that authentic learning and growth often occur when one steps outside one's comfort zone. This approach is particularly relevant in a field that combines biology's complexities with computational methods' challenges. As you navigate this course, you will encounter concepts, problems, and methodologies that may initially seem daunting or unfamiliar. This is intentional. By pushing the boundaries of your knowledge and skills, you develop resilience, adaptability, and a deeper understanding of the subject matter.
Rest assured that your willingness to step outside your comfort zone will not negatively affect your grade. We have meticulously structured the course to support your growth while ensuring fair and transparent assessment methods.
- Most assignments employ a scaffolded or tiered approach, where more straightforward questions that test foundational knowledge are worth more points than the more challenging, advanced questions.
- Optional "stretch" questions or projects for those seeking extra challenges, with bonus points that can't negatively affect your grade.
These strategies are designed to create a nurturing and supportive learning environment where you can push your boundaries, take intellectual risks, and grow your skills in computational biology without fear of academic consequences.
Real-world scenarios enhance learning¶
As we delve into computational biology, I want to emphasize the practical relevance of each module. Instead of a traditional approach, we'll use motivating real-world scenarios that underscore the importance of the concepts you'll be learning. Consider yourself a problem solver in a scientific expedition, applying computational tools to tackle actual challenges faced in the field. Our focus will be on tangible applications, from predicting the impact of genetic variations on disease susceptibility to simulating the dynamics of biological systems under different conditions. This approach ensures that what you learn in this course is not confined to theoretical frameworks but has direct implications for understanding and addressing complex biological phenomena.
Connecting you to opportunities¶
Embarking on a journey in computational biology is most effectively achieved by actively participating in a research lab at Pitt. This hands-on experience enriches your understanding and provides a practical immersion; computational biology research is becoming an expectation to remain competitive in today's job market. The modules in this course integrate seamlessly with this approach, offering direct links to labs that leverage tools relevant to the specific content covered. You should contact these labs to see if they have paid or for-credit research positions available.
Single source of truth¶
Each course should have a single source of truth (SSOT), which is having a single authoritative source of data and information across the course. Having an SSOT can help you access all the information related to the course in one place. SSOT can help reduce confusion and ensure everyone is on the same page. For this course, the SSOT is pitt-biosc1540-2024f.oasci.org.
Another benefit of having an SSOT website is that it can help keep the course content up-to-date and accurate. Your instructor can change the website as needed; you will always have access to the most current information. This can help ensure you learn the most relevant and up-to-date material. Finally, having an SSOT website can help improve communication between your instructor and the students. Your instructor can share important announcements, assignments, and other course-related information on the website. This can help ensure everyone is informed and up-to-date on what's happening in the course.
Everything is a draft¶
Open science and education are my core values and extend to my teaching. You will likely see me working on things I assign in an hour or next week. Teaching new courses or making significant changes always works like this. Thus, looking ahead is acceptable, and all drafts are marked with the following admonition. If you start working ahead and things change, your time is lost.
DRAFT
This page is a work in progress and is subject to change at any moment.
I will do my best to avoid mistakes, but they can happen. If you see grammar or spelling issues or need clarification on passages that don't have the DRAFT admonition, feel free to tell me so I can correct them.
Outcomes¶
There are only two required courses in the Bachelor's degree in computational biology where learning computational biology is the focus. After this course, students must take computational genomics (BIOSC 1542) or simulation and modeling (BIOSC 1544). Outcomes for this course are geared towards introducing students to both subfields.
- Understand the fundamental concepts and methodologies in genomics, transcriptomics, and computational structural biology.
- Recognize the applications of computational methods in various biological fields.
- Critically evaluate the strengths and limitations of different computational approaches in biology.
- Interpret and analyze results from various computational biology methods.
- Demonstrate enhanced problem-solving and critical thinking skills in the context of computational biology.
Schedule¶
We start with bioinformatics to learn fundamentals of genomics and transcriptomics. Afterwards, we will cover structure prediction, molecular simulations, and computer-aided drug design in our computational structural biology module. Our third module in scientific python will introduce how we can use Python in computational biology.
Module 01 - Bioinformatics¶
Week 1¶
Tuesday (Aug 27) Lecture 01
- Topics: Introduction to computational biology.
Thursday (Aug 29) Lecture 02
- Topics: DNA sequencing technologies.
Week 2¶
Tuesday (Sep 3) Lecture 03
- Topics: Quality control.
Thursday (Sep 5) Lecture 04
- Topics: De novo genome assembly.
Week 3¶
Tuesday (Sep 10) Lecture 05
- Topics: Gene annotation.
Thursday (Sep 12) Lecture 06
- Topics: Sequence alignment.
Week 4¶
Tuesday (Sep 17) Lecture 07
- Topics: Introduction to transcriptomics.
Thursday (Sep 19) Lecture 08
- Topics: Read mapping.
Week 5¶
Tuesday (Sep 24) Lecture 09
- Topics: Gene expression quantification.
Thursday (Sep 26) Lecture 10
- Topics: Differential gene expression.
Week 6¶
Tuesday (Oct 1)
- Topics: Review
Thursday (Oct 3)
- Topics: Exam 1
Module 02 - Computational structural biology¶
Week 7¶
Tuesday (Oct 8) Lecture 11
- Topics: Structural biology and determination.
Thursday (Oct 10) Lecture 12
- Topics: Protein structure prediction.
Week 8¶
Tuesday (Oct 15) Fall break (no class)
Thursday (Oct 17) Lecture 13
- Topics: Molecular simulation principles.
Week 9¶
Tuesday (Oct 22) Lecture 14
- Topics: Molecular system representations.
Thursday (Oct 24) Lecture 15
- Topics: Gaining atomistic insights.
Week 10¶
Tuesday (Oct 29) Lecture 16
- Topics: Structure-based drug design.
Thursday (Oct 31) Lecture 17
- Topics: Docking and virtual screening.
Week 11¶
Tuesday (Nov 5) Election day: Go vote (no class)
Thursday (Nov 7) Lecture 18
- Topics: Ligand-based drug design.
Week 12¶
Tuesday (Nov 12) Review
Thursday (Nov 14): Exam 2
Module 03 - Scientific python¶
These are optional lectures that will not negatively affect your final grade.
Week 13¶
Tuesday (Nov 19) Lecture 19
- Topics: Python basics
Thursday (Nov 21) Lecture 20
- Topics: Arrays and plotting
Thanksgiving break¶
No class on Nov 26 and 28.
Week 15¶
Tuesday (Dec 3) Lecture 21
- Topics: Predictive modeling
Thursday (Dec 5) Lecture 22
- Topics: Project work
Week 16¶
Tuesday (Dec 10) Lecture 23
- Topics: Project work
Final¶
Monday (Dec 16) Final exam
- An optional, cumulative final exam will be offered to replace lowest exam grade.
Assessments¶
Distribution¶
The course will have the following point distribution.
- Exams (35%)
- Project (30%)
- Assignments (35%)
Late assignments and extensions¶
I am mindful of the diverse nature of deadlines, particularly in the scientific realm. Some are set in stone, while others exhibit more flexibility. It is noteworthy that the scientific community frequently submits manuscripts and reviews days, weeks, or months after the editor's request. Such practices are widely understood. Conversely, submitting a grant application even a minute past the deadline makes it ineligible for review.
I will use the following late assignment and extension policy. It encourages timely submissions while acknowledging the influence of external commitments and unforeseen circumstances.
- Each assignment has a specified due date and time.
- Assignments submitted after the due date will incur a late penalty.
-
The late penalty is calculated using the function: % Penalty = 0.01 (1.4 \(\times\) hours late)2 rounded to the nearest tenth. This results in approximately:
Hours late Penalty 6 0.7% 12 2.8% 24 11.3% 48 45.2% 72 100.0% -
Assignments will not be accepted more than 72 hours (3 days) after the due date.
- The penalty is applied to the assignment's total possible points. For example, if an assignment is worth 100 points and is submitted 36 hours late, the penalty would be approximately 13 points.
- To reward punctuality, each assignment submitted on time will earn a 2% bonus on that assignment's score.
- These on-time bonuses will accumulate throughout the semester and will be added to your final course grade.
Exceptions to this policy will be made on a case-by-case basis for extenuating circumstances. Please communicate with me as early as possible if you anticipate difficulties meeting a deadline.
Submitting your assignments on time can earn you up to a 2% bonus added to your final grade. For each assignment you submit on time, you will receive a proportional percentage of this bonus. For example, if you submit 7 out of 8 assignments on time, you would receive a 1.75% boost to your final grade. This bonus can help improve your final grade, but please note that I will not round up final grades.
Missed exams¶
Attendance and participation in all scheduled exams are crucial for your success in this course. To accommodate unforeseen circumstances while maintaining academic standards, please be aware of the following policy:
Due to time constraints and the difficulty of creating equivalent assessments, makeup exams will only be offered under the same circumstances. Instead, an optional cumulative final exam will be available at the end of the course. This final exam can replace your lowest midterm exam grade if it benefits your overall score. If the final exam score is lower than your existing midterm grades, it will not be counted, ensuring it cannot negatively impact your grade.
If you miss one of the midterm exams, you are highly encouraged to take the cumulative final exam. The score you achieve on the final exam will replace the missed midterm exam grade in calculating your final grade. Missing both midterm exams poses a significant challenge to course completion; in such cases, you must contact me immediately to discuss your situation and explore possible options.
Scale¶
Letter grades for this course will be assigned based on Pitt's recommended scale (shown below).
| Letter grade | Percentage | GPA |
|---|---|---|
| A + | 97.0 - 100.0% | 4.00 |
| A | 93.0 - 96.9% | 4.00 |
| A – | 90.0 - 92.9% | 3.75 |
| B + | 87.0 - 89.9% | 3.25 |
| B | 83.0 - 86.9% | 3.00 |
| B – | 80.0 - 82.9% | 2.75 |
| C + | 77.0 - 79.9% | 2.25 |
| C | 73.0 - 76.9% | 2.20 |
| C – | 70.0 - 72.9% | 1.75 |
| D + | 67.0 - 69.9% | 1.25 |
| D | 63.0 - 66.9% | 1.00 |
| D – | 60.0 - 62.9% | 0.75 |
| F | 0.0 - 59.9% | 0.00 |
Attendance
Attendance and participation¶
While attendance is not mandatory, active participation is strongly encouraged and integral to your course success. Regular class attendance offers several benefits:
- Enhanced understanding of course material through engaging discussions;
- Opportunities for collaborative learning through group activities;
- Practical application of concepts via hands-on exercises.
Please note that while it is possible to achieve the maximum number of points without attending lectures, consistent attendance demonstrates commitment to the course. This commitment may be considered when addressing individual circumstances or requests for flexibility.
Important
Virtual attendance (e.g., via Zoom) is unavailable for this course. All class sessions will be conducted in person.
Policies¶
Generative AI¶
We are in an exciting area of generative AI development with the release of tools such as ChatGPT, DALL-E, GitHub Copilot, Bing Chat, Bard, Copy.ai, and many more. This course will permit these tools' ethical and responsible use except when explicitly noted. For example, you can use these tools as an on-demand tutor by explaining complex topics.
Other ways are undoubtedly possible, but any use should aid—not replace—your learning. You must also be aware of the following aspects of generative AI.
-
AI limitations: While AI programs can be valuable resources, they may produce inaccurate, biased, or incomplete material. Each program has its unique limitations as well.
-
Bias and accuracy: Scrutinizing each aspect of these enormous data sets used to train these products is infeasible. AI will inherit biases and inaccuracies from these sources and human influences in fine-tuning. You must be critical and skeptical of anything generated from these models and verify information from trusted sources.
-
Critical thinking: Understand that AI is a tool, not a replacement for your analysis and critical thinking skills. AI to enhance your understanding and productivity, but remember that your development as a scholar depends on your ability to engage independently with the material.
-
Academic integrity: Plagiarism extends to content generated by AI. Using AI-generated material without proper attribution is a violation of academic integrity policies. Always give credit to AI-generated content and adhere to citation rules.
Furthermore, text from AI tools should be treated as someone else's work—because it is. You should never copy and paste text directly.
-
AI detection: As discussed here, the University Center for Teaching and Learning does not recommend using AI detection tools like turnitin due to high false positive rates. I will not use AI detection tools in any capacity for this course and trust that you will use these tools responsibly when permitted and desired.
Remember that generative AI is helpful when used responsibly. You can ethically benefit from these technological advances by adhering to these guidelines. Embrace this opportunity to expand your skill set and engage thoughtfully with emerging technologies. If you have any questions about AI tool usage, please get in touch with me for clarification and guidance.
Equity, diversity, and inclusion¶
The University of Pittsburgh does not tolerate any form of discrimination, harassment, or retaliation based on disability, race, color, religion, national origin, ancestry, genetic information, marital status, familial status, sex, age, sexual orientation, veteran status or gender identity or other factors as stated in the University's Title IX policy. The University is committed to taking prompt action to end a hostile environment that interferes with the University's mission. For more information about policies, procedures, and practices, visit the Civil Rights & Title IX Compliance web page.
I ask that everyone in the class strive to help ensure that other members of this class can learn in a supportive and respectful environment. If there are instances of the aforementioned issues, please contact the Title IX Coordinator, by calling 412-648-7860 or emailing titleixcoordinator@pitt.edu. Reports can also be filed online. You may also choose to report this to a faculty/staff member; they are required to communicate this to the University's Office of Diversity and Inclusion. If you wish to maintain complete confidentiality, you may also contact the University Counseling Center (412-648-7930).
Academic integrity¶
Students in this course will be expected to comply with the University of Pittsburgh's Policy on Academic Integrity. Any student suspected of violating this obligation during the semester will be required to participate in the procedural process initiated at the instructor level, as outlined in the University Guidelines on Academic Integrity. This may include, but is not limited to, the confiscation of the examination of any individual suspected of violating University Policy. Furthermore, no student may bring unauthorized materials to an exam, including dictionaries and programmable calculators.
To learn more about Academic Integrity, visit the Academic Integrity Guide for an overview. For hands-on practice, complete the Understanding and Avoiding Plagiarism tutorial.
Disability services¶
If you have a disability for which you are or may be requesting an accommodation, you are encouraged to contact both your instructor and Disability Resources and Services (DRS), 140 William Pitt Union, (412) 648-7890, drsrecep@pitt.edu, (412) 228-5347 for P3 ASL users, as early as possible in the term.
DRS will verify your disability and determine reasonable accommodations for this course.
Email communication¶
Upon admittance, each student is issued a University email address (username@pitt.edu).
The University may use this email address for official communication with students.
Students are expected to read emails sent to this account regularly.
Failure to read and react to University communications promptly does not absolve the student from knowing and complying with the content of the communications.
The University provides an email forwarding service that allows students to read their email via other service providers (e.g., Gmail, AOL, Yahoo).
Students who forward their email from their pitt.edu address to another address do so at their own risk.
If email is lost due to forwarding, it does not absolve the student from responding to official communications sent to their University email address.
Religious observances¶
The observance of religious holidays (activities observed by a religious group of which a student is a member) and cultural practices are an important reflection of diversity. As your instructor, I am committed to providing equivalent educational opportunities to students of all belief systems. At the beginning of the semester, you should review the course requirements to identify foreseeable conflicts with assignments, exams, or other required attendance. If possible, please contact me (your course coordinator/s) within the first two weeks of the first class meeting to allow time for us to discuss and make fair and reasonable adjustments to the schedule and/or tasks.
Sexual misconduct, required reporting, and Title IX¶
If you are experiencing sexual assault, sexual harassment, domestic violence, and stalking, please report it to me and I will connect you to University resources to support you.
University faculty and staff members are required to report all instances of sexual misconduct, including harassment and sexual violence to the Office of Civil Rights and Title IX. When a report is made, individuals can expect to be contacted by the Title IX Office with information about support resources and options related to safety, accommodations, process, and policy. I encourage you to use the services and resources that may be most helpful to you.
As your instructor, I am required to report any incidents of sexual misconduct that are directly reported to me. You can also report directly to Office of Civil Rights and Title IX: 412-648-7860 (M-F; 8:30am-5:00pm) or via the Pitt Concern Connection at: Make A Report.
An important exception to the reporting requirement exists for academic work. Disclosures about sexual misconduct that are shared as a relevant part of an academic project, classroom discussion, or course assignment, are not required to be disclosed to the University's Title IX office.
If you wish to make a confidential report, Pitt encourages you to reach out to these resources:
- The University Counseling Center: 412-648-7930 (8:30 A.M. TO 5 P.M. M-F) and 412-648-7856 (AFTER BUSINESS HOURS)
- Pittsburgh Action Against Rape (community resource): 1-866-363-7273 (24/7)
If you have an immediate safety concern, please contact the University of Pittsburgh Police, 412-624-2121
Any form of sexual harassment or violence will not be excused or tolerated at the University of Pittsburgh.
For additional information, please visit the full syllabus statement on the Office of Diversity, Equity, and Inclusion webpage.
Statement on classroom recording¶
To ensure the free and open discussion of ideas, students may not record classroom lectures, discussions and/or activities without the advance written permission of the instructor, and any such recording properly approved in advance can be used solely for the student's private use.
Ended: Syllabus
Lectures ↵
Lectures¶
We have 22 lectures spanned across three modules:
Bioinformatics ↵
Bioinformatics¶
In this module, we cover experimental and computational foundations of genomics and transcriptomics.
Lecture 01
Computational biology course overview and foundations
Date: Aug 27, 2024
This inaugural lecture serves as a comprehensive introduction to the field of computational biology and establishes the framework for the course.
Learning objectives¶
What you should be able to do after today's lecture:
- Describe the course structure, expectations, and available resources for success.
- Define computational biology and explain its interdisciplinary nature.
- Identify key applications and recent advancements.
- Understand the balance between applications and development.
- Identify potential career paths and educational opportunities.
Presentation¶
Download: biosc1540-l01.pdf
Lecture 02
DNA sequencing
Date: August 29, 2024
The session traces the evolution of DNA sequencing technologies, commencing with the seminal Sanger method and progressing to state-of-the-art Illumina and Nanopore platforms. Through a comparative analysis of these technologies, we will elucidate the rapid advancements that have catalyzed genomic research. The discourse will encompass a critical evaluation of the strengths and limitations inherent to each method, offering insights into their optimal applications in contemporary research contexts. The lecture culminates with an in-depth overview of a typical DNA sequencing workflow, providing students with a pragmatic understanding of genomic data generation.
Learning objectives¶
What you should be able to do after today's lecture:
- Analyze antibiotic resistance challenges and evaluate genomics' role in mitigating them.
- Construct a general workflow intrinsic to DNA sequencing experiments.
- Delineate the core principles underlying Sanger sequencing.
- Conduct a comparative analysis of Illumina sequencing vis-à-vis Sanger sequencing.
- Explicate the fundamental principles governing Nanopore sequencing technology.
Readings¶
Relevant content for today's lecture.
- Sequencing background
- DNA sequencing
- DNA sample preparation
- PCR
- Sanger sequencing
- Illumina sequencing
- Nanopore sequencing
Presentation¶
Download: biosc1540-l02.pdf
Lecture 03: Quality control ↵
Lecture 03
Quality control
Date: Sep 3, 2024
This session focuses on the critical steps of quality control and preprocessing in genomic data analysis. We will explore the fundamentals of sequencing data formats and delve into essential techniques for assessing and improving data quality.
Learning objectives¶
What you should be able to do after today's lecture:
- Explain the basic concepts and importance of genome assembly.
- Interpret FASTA and FASTQ file formats and their role in storing sequences.
- Perform and interpret quality control on reads using FastQC.
- Identify common quality issues in sequencing data and explain their impacts.
- Describe the process and importance of sequence trimming and filtering.
Readings¶
Relevant content for today's lecture.
- Genome assembly
- Assembly concepts and nested content
- FASTA files
- FASTQ files
- FastQC and nested content
Presentation¶
Download: biosc1540-l03.pdf
Activities ↵
Ended: Activities
Ended: Lecture 03: Quality control
Lecture 04
De novo genome assembly
Date: Sep 5, 2024
This lecture aims to provide a thorough understanding of de novo assembly techniques, their applications, and their crucial role in advancing genomic research.
Learning objectives¶
What you should be able to do after today's lecture:
- Explain the fundamental challenge of reconstructing a complete genome.
- Describe and apply the principles of the greedy algorithm.
- Understand and construct de Bruijn graphs.
Readings¶
Relevant content for today's lecture.
- Greedy algorithm
- de Bruijn
- JHU
- JHU2
- Why are de Bruijn graphs useful for genome assembly?
- Read mapping on de Bruijn graphs
- Galaxy
- Data science for HTS
- SPAdes assembler
- Online graph builder. Note that they use knode values.
Presentation¶
Download: biosc1540-l04.pdf
Lecture 05
Gene annotation
Date: Sep 10, 2024
This lecture covers the fundamental concepts and techniques of gene annotation, a critical process in genomics for identifying and characterizing genes within DNA sequences. We'll explore various computational methods used in gene annotation, their applications, and challenges.
Learning Objectives¶
By the end of this lecture, you should be able to:
- Explain the graph traversal and contig extraction process in genome assemblers.
- Understand key output files and quality metrics of genome assembly results.
- Define gene annotation and describe its key components.
- Outline the main computational methods used in gene prediction and annotation.
- Analyze and interpret basic gene annotation data and outputs.
Readings¶
Relevant content for today's lecture.
- None! Just the lecture.
Presentation¶
Download: biosc1540-l05.pdf
Lecture 06
Sequence alignment
Date: Sep 12, 2024
In this lecture, we will explore sequence alignment, a fundamental technique in computational biology. We will cover the basics of alignment scoring, key algorithms for both pairwise and multiple sequence alignments, and learn how to interpret alignment results.
Learning objectives¶
What you should be able to do after today's lecture:
- Define sequence alignment and explain its importance in bioinformatics.
- Describe the basic principles of scoring systems in sequence alignment.
- Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
- Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
- Introduce the concept of multiple sequence alignment, including its importance and challenges.
Readings¶
Relevant content for today's lecture.
- Global alignment
- Local alignment
- Online tools for Needleman-Wunsch and Smith-Waterman.
Presentation¶
Download: biosc1540-l06.pdf
Lecture 07
Introduction to transcriptomics
Date: Sep 17, 2024
By the end of this lecture, you'll have a comprehensive understanding of transcriptomics' power in unraveling the intricacies of gene expression and its broad applications in biological research.
Learning objectives¶
What you should be able to do after today's lecture:
- Define transcriptomics and explain its role in understanding gene expression patterns.
- Discuss emerging trends in transcriptomics.
- Compare and contrast transcriptomics and genomics.
- Explain the principles of RNA-seq technology and its advantages over previous methods.
- Outline the computational pipeline for RNA-seq data analysis.
Readings¶
Relevant content for today's lecture.
- None! Just the presentation.
Presentation¶
Download: biosc1540-l07.pdf
Lecture 08
Read mapping
Date: Sep 19, 2024
We'll explore the challenges of aligning millions of short reads to a reference genome and discuss various algorithms and data structures that make this process efficient. The session will focus on the Burrows-Wheeler Transform (BWT) and the FM-index, two key concepts that revolutionized read alignment by enabling fast, memory-efficient sequence searching. We'll examine how these techniques are implemented in popular alignment tools and compare their performance characteristics.
Learning objectives¶
What you should be able to do after today's lecture:
- Describe the challenges of aligning short reads to a large reference genome.
- Compare read alignment algorithms, including hash-based and suffix tree-based approaches.
- Explain the basic principles of the Burrows-Wheeler Transform (BWT) for sequence alignment.
Readings¶
Relevant content for today's lecture.
- Burrows-Wheeler transform
- Suffix trees
- Suffix arrays
Presentation¶
Download: biosc1540-l08.pdf
Lecture 09
Gene expression quantification
Date: Sep 24, 2024
We'll explore how raw sequencing reads are transformed into meaningful measures of gene activity, navigating the complexities of multi-mapped reads and isoform variations. The session will compare various quantification metrics, from traditional RPKM to more recent innovations like TPM, highlighting their strengths and limitations. We'll examine cutting-edge tools for transcript-level quantification and discuss the crucial role of normalization in generating comparable expression data across samples. Through practical examples, students will learn to interpret gene expression results, bridging the gap between computational output and biological insight.
Learning objectives¶
What you should be able to do after today's lecture:
- Discuss the importance of normalization and quantification in RNA-seq data analysis.
- Explain the relevance of pseudoalignment instead of read mapping.
- Understand the purpose of Salmon's generative model.
- Describe how salmon handles experimental biases in transcriptomics data.
- Communicate the principles of inference in Salmon.
Readings¶
Relevant content for today's lecture.
Presentation¶
Download: biosc1540-l09.pdf
Lecture 10
Differential gene expression
Date: Sep 26, 2024
This lecture explores differential gene expression analysis, a powerful approach for uncovering the molecular basis of biological phenomena.
Learning objectives¶
What you should be able to do after today's lecture:
- Define differential gene expression and explain its importance.
- Describe the statistical principles underlying differential expression analysis.
- Outline the steps in a typical differential expression analysis workflow.
- Explain key concepts such as fold change, p-value, and false discovery rate.
- Interpret common visualizations used in differential expression analysis.
Readings¶
Relevant content for today's lecture.
- None! Just the slides.
Presentation¶
Download: biosc1540-l10.pdf
Ended: Bioinformatics
Computational structural biology ↵
Computational structural biology¶
In this module, we cover computational structural biology.
Lecture 11
Structural biology
Date: Oct 8, 2024
This lecture introduces the field of computational structural biology and its interplay with experimental methods. We'll explore how computational approaches complement and enhance traditional structural biology techniques, providing insights into protein structure, function, and dynamics. The session will cover key experimental methods like X-ray crystallography and cryo-EM, highlighting their strengths and limitations.
Learning objectives¶
What you should be able to do after today's lecture:
- Categorize atomic interactions and their importance.
- Explain what is structural biology and why is it important.
- Communicate the basics of X-ray crystallization.
- Find and analyze protein structures in the Protein Data Bank (PDB).
- Compare and contrast Cryo-EM to X-ray crystallization.
- Understand the experimental challenges of disorder.
- Know why protein structure prediction is helpful.
Readings¶
Relevant content for today's lecture.
- None! Just the lecture.
Presentation¶
Download: biosc1540-l11.pdf
Lecture 12
Protein structure prediction
Date: Oct 10, 2024
We'll explore how amino acid sequences are transformed into three-dimensional structures through computational methods. The session will cover various approaches, from traditional homology modeling to cutting-edge deep learning techniques like AlphaFold. We'll examine the principles underlying these methods, their applications, and their impact on biological research.
Learning objectives¶
What you should be able to do after today's lecture:
- Why are we learning about protein structure prediction?
- Identify what makes structure prediction challenging.
- Explain homology modeling.
- Know when to use threading instead of homology modeling.
- Interpret a contact map for protein structures.
- Comprehend how coevolution provides structural insights.
- Explain why ML models are dominate protein structure prediction.
Readings¶
Relevant content for today's lecture.
- None! Just the lecture.
Presentation¶
Download: biosc1540-l12.pdf
Lecture 13
Molecular simulation principles
Date: Oct 17, 2024
This lecture introduces the fundamental principles of molecular dynamics (MD) simulations. We'll explore the theoretical foundations of MD, including force fields and integration algorithms. The session will cover the basic concepts needed to understand how MD simulations provide a dynamic view of molecular systems, complementing static structural data.
Learning objectives¶
What you should be able to do after today's lecture:
- Understand the importance of molecular dynamics (MD) simulations for proteins.
- Identify the validity of the classical approximation.
- Discuss the concept of equations of motion in MD simulations.
- Explain the role of integration algorithms in MD simulations.
- Describe the components of a molecular mechanics force field.
- Understand noncovalent contributions to force fields.
- Identify data for force field parameterization
Readings¶
Relevant content for today's lecture.
- None! Just the slides (and Google).
Presentation¶
Download: biosc1540-l13.pdf
Lecture 14
Molecular system representations
Date: Oct 22, 2024
Building on the foundations from the previous lecture, this session focuses on the practical aspects of setting up and running MD simulations. We'll walk through the steps involved in preparing a system for simulation.
Note
I decided to go over MD relaxations a second time for L15 and moved those slides there.
Learning objectives¶
What you should be able to do after today's lecture:
- Explain why DHFR is a promising drug target.
- Select and prepare a protein structure for molecular simulations.
- Explain the importance of approximating molecular environments.
- Describe periodic boundary conditions and their role in MD simulations.
- Explain the role of force field selection and topology generation.
- Outline the process of energy minimization and its significance.
Readings¶
Relevant content for today's lecture if you are interested.
Presentation¶
Download: biosc1540-l14.pdf
Lecture 15
Ensembles and atomistic insights
Date: Oct 24, 2024
This final lecture in the MD series focuses on the analysis and interpretation of MD simulation data. We'll explore common analysis techniques and how to extract meaningful biological insights from simulation trajectories.
Learning objectives¶
After today, you should better understand:
- Molecular ensembles and their relevance.
- Maintaining thermodynamic equilibrium.
- Relaxation and production MD simulations.
- RMSD and RMSF as conformational changes and flexibility metrics.
- Relationship between probability and energy in simulations.
Readings¶
Relevant content for today's lecture.
- Any scientific literature.
Presentation¶
Download: biosc1540-l15.pdf
Lecture 16
Structure-based drug design
Date: Oct 29, 2024
What you should be able to do after today's lecture:
- Drug development pipeline.
- Role of structure-based drug design.
- Thermodynamics of binding.
- Enthalpic contributions to binding.
- Entropic contributions to binding
- Alchemical free energy simulations
Presentation¶
Download: biosc1540-l16.pdf
Lecture 17
Docking and virtual screening
Date: Oct 31, 2024
This lecture delves into molecular docking and virtual screening, essential techniques in computational drug discovery. The session will cover different docking algorithms, scoring functions, and strategies for improving virtual screening efficiency and accuracy.
Learning objectives¶
After today, you should have a better understanding of:
- Practical limitations of alchemical simulations.
- Value of data-driven approaches for docking.
- Identifying relevant protein conformations.
- Detecting binding pockets.
- Ligand pose optimization.
- Scoring functions as data-driven predictors.
- Interpretation of docking results.
Presentation¶
Download: biosc1540-l17.pdf
Lecture 18
Ligand-based drug design
Date: Nov 7, 2024
After today, you should have a better understanding of:
- The basic principles of ligand-based drug design and how it differs from structure-based approaches.
- How descriptors and fingerprints evaluate molecular similarity.
- How QSAR models predict biological activity based on molecular structure.
- The role of pharmacophore modeling in identifying essential molecular features for activity.
Presentation¶
Download: biosc1540-l18.pdf
Ended: Computational structural biology
Scientific python ↵
Scientific python¶
These are optional lectures will teach you the fundamentals of scientific Python. Not attending these lectures will not impact your grade.
Lecture 19
Python basics
Date: Nov 19, 2024
Learning Objectives¶
After today, you should have a better understanding of:
- Setting up and navigating a Colab notebook to write and execute Python code.
- Writing simple print statements to display output.
- Adding inline and block comments to make code more understandable.
- Using Python to perform basic arithmetic calculations like addition, subtraction, multiplication, division, and exponentiation.
- Defining and assigning variables.
- Recognizing and using basic Python data types such as integers, floats, strings, and booleans.
- Implementing decision-making logic using
if,elif, andelsestatements. - Using lists, tuples, and dictionaries to store and manipulate grouped data.
- Writing reusable code by defining functions with parameters and return values.
- Iterating over sequences using
forloops. - Writing
whileloops to repeat actions based on conditions.
Materials¶
Additional resources¶
- Here are some additional resources for learning Python: kaggle learn, Software Carpentry, learnpython.org, and Google's Python Class
Lecture 20
Arrays
Date: Nov 21, 2024
Learning objectives¶
What you should be able to do after today's lecture:
- Learn how to load, slice, and manipulate multi-dimensional data arrays.
- Calculate descriptive statistics such as mean, median, and standard deviation.
- Generate scatter plots to visualize relationships between variables.
- Use heat maps to explore gene expression patterns across patients.
- Compare gene expression levels between survivors and non-survivors.
- Identify potential genes of interest using a volcano plot.
Materials¶
Additional resources¶
- Here are some additional resources for learning Python: python.crumblearn.org/data/numpy/
Lecture 21: Predictive modeling ↵
Lecture 21
Predictive modeling
Date: Dec 3, 2024
Learning objectives¶
After today, you should have a better understanding of:
- Define linear regression, its limitations, and objective function.
- Describe the purpose of loss functions in regression.
- Understand the conversion of data from a DataFrame to NumPy arrays.
- Develop hands-on programming skills for implementing regression in Python.
- Interpret the coefficients obtained through optimization and evaluate the model's performance.
- Visualize linear regression models and their fit to data.
- Discuss practical considerations for model interpretation, assumptions, and limitations.
Some relevant code snippets.
import numpy as np
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
import py3Dmol
def show_mol(smi, style="stick"):
mol = Chem.MolFromSmiles(smi)
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol, maxIters=200)
mblock = Chem.MolToMolBlock(mol)
view = py3Dmol.view(width=500, height=500)
view.addModel(mblock, "mol")
view.setStyle({style: {}})
view.zoomTo()
view.show()
Predictive Modeling - pKa edition¶
Today, we delve into the application of regression analysis in chemistry and data science. Our dataset comprises p$K_\text{a}$ values and corresponding molecular descriptors, offering a quantitative approach to understanding molecular properties.
pKa¶
The p$K_\text{a}$ measures a substance's acidity or basicity, particularly in chemistry. It is the negative logarithm (base 10) of the acid dissociation constant ($K_\text{a}$) of a solution. The p$K_\text{a}$ value helps quantify the strength of an acid in a solution.
The expression for the acid dissociation constant ($K_\text{a}$), from which p$K_\text{a}$ is derived, is given by the following chemical equilibrium equation for a generic acid (HA) in water:
$$ \text{HA} \rightleftharpoons \text{H}^+ + \text{A}^- $$
The equilibrium constant ($K_\text{a}$) for this reaction is defined as the ratio of the concentrations of the dissociated ions ($\text{H}^+$ and $\text{A}^-$) to the undissociated acid ($\text{HA}$):
$$ K_a = \frac{[\text{H}^+][\text{A}^-]}{[\text{HA}]} $$
Taking the negative logarithm (base 10) of both sides of the equation gives the expression for pKa:
$$ \text{p}K_a = -\log_{10}(K_a) $$
So, in summary, the p$K_\text{a}$ is calculated by taking the negative logarithm of the acid dissociation constant ($K_\text{a}$) for a given acid. A lower p$K_\text{a}$ indicates a stronger acid.
In simpler terms:
- A lower p$K_\text{a}$ indicates a stronger acid because it means the acid is more likely to donate a proton (H+) in a chemical reaction.
- A higher p$K_\text{a}$ indicates a weaker acid as it is less likely to donate a proton.
The p$K_\text{a}$ is a crucial parameter in understanding the behavior of acids and bases in various chemical reactions. It is commonly used in fields such as medicinal chemistry, biochemistry, and environmental science to describe and predict the behavior of molecules in solution.
Exploring the dataset¶
Now, let's shift our focus to a practical application of our theoretical knowledge.
I found this dataset that contains a bunch of high-quality experimental measurements of pKas. It contains a bunch of information and other aspects that makes regression a bit of a nightmare; thus, I did some cleaning of the data and computed some molecular features (i.e., descriptors) that we can use. Before delving into regression analysis, it is essential to conduct a systematic review of the dataset. This preliminary examination will provide us with the necessary foundation to understand the quantitative relationships between molecular features and acidity. Let's now proceed with a methodical investigation of the empirical data, setting the stage for our subsequent analytical endeavors.
Loading¶
Using the Pandas library, read the CSV file into a DataFrame. Use the variable you defined in the previous step.
import numpy as np
import pandas as pd
# @title
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
import py3Dmol
def show_mol(smi, style="stick"):
mol = Chem.MolFromSmiles(smi)
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol, maxIters=200)
mblock = Chem.MolToMolBlock(mol)
view = py3Dmol.view(width=500, height=500)
view.addModel(mblock, "mol")
view.setStyle({style: {}})
view.zoomTo()
view.show()
CSV_PATH = "https://github.com/oasci-courses/pitt-biosc1540-2024f/raw/refs/heads/main/content/lectures/21/smiles-pka-desc.csv"
df = pd.read_csv(CSV_PATH)
Ended: Lecture 21: Predictive modeling
Lecture 22
Project work
Date: Dec 5, 2024
For today's lecture, we will have an open work session dedicated to your ongoing projects. This is a great opportunity to focus on your work, collaborate with classmates, and address any challenges you might be facing. Additionally, I will be available to provide help with PyMol, a powerful molecular visualization tool that can enhance your project presentations and analyses.
Lecture 23
Project work
Date: Dec 10, 2024
For today's lecture, we will have an open work session dedicated to your ongoing projects. This is a great opportunity to focus on your work, collaborate with classmates, and address any challenges you might be facing. Additionally, I will be available to provide help with PyMol, a powerful molecular visualization tool that can enhance your project presentations and analyses.
Ended: Scientific python
Ended: Lectures
Assessments ↵
Assessments¶
Welcome to the assessments page for BIOSC 1540 - Computational Biology. This page provides an overview of how your performance in the course will be evaluated.
Distribution¶
Your final grade will be calculated based on the following distribution:
- Exams (35%)
- Project (30%)
- Assignments (35%)
Grading Scale¶
Letter grades will be assigned based on the following scale:
| Letter grade | Percentage | GPA |
|---|---|---|
| A + | 97.0 - 100.0% | 4.00 |
| A | 93.0 - 96.9% | 4.00 |
| A – | 90.0 - 92.9% | 3.75 |
| B + | 87.0 - 89.9% | 3.25 |
| B | 83.0 - 86.9% | 3.00 |
| B – | 80.0 - 82.9% | 2.75 |
| C + | 77.0 - 79.9% | 2.25 |
| C | 73.0 - 76.9% | 2.20 |
| C – | 70.0 - 72.9% | 1.75 |
| D + | 67.0 - 69.9% | 1.25 |
| D | 63.0 - 66.9% | 1.00 |
| D – | 60.0 - 62.9% | 0.75 |
| F | 0.0 - 59.9% | 0.00 |
For more detailed information on assessments, please consult the full course syllabus or contact the instructor.
Assignments ↵
Assignments¶
We will have seven homework assignments throughout the semester.
A01
Please submit your answers as a PDF to gradescope.
Q01¶
Points: 6
Why is the quality of sequencing data typically lower at the end of a read in Sanger sequencing?
Solution
The quality of sequencing data in Sanger sequencing tends to decrease towards the end of a read due to several factors related to the nature of the sequencing process. This phenomenon is primarily attributed to the decreasing population of longer DNA fragments. Here's a detailed explanation:
- Probability of ddNTP incorporation:
- To obtain longer fragments, we need to avoid incorporating a dideoxynucleotide (ddNTP) until the very end of the sequence.
- As the length of the fragment increases, the probability of not incorporating a ddNTP at any previous position decreases.
- This results in fewer long fragments compared to shorter ones, leading to weaker signals for longer sequences.
- Concentration ratio of dNTPs to ddNTPs:
- While we maintain a low concentration of ddNTPs relative to dNTPs to promote the synthesis of longer fragments, the cumulative probability of ddNTP incorporation still increases with length.
- This leads to a gradual decrease in the population of fragments as their length grows.
- Mass and mobility differences:
- As DNA fragments increase in length, the relative mass difference between fragments of consecutive lengths decreases.
- For example, the mass difference between a 99-mer and a 100-mer is proportionally smaller than the difference between a 9-mer and a 10-mer.
- This results in poorer separation of longer fragments during electrophoresis, contributing to decreased resolution and quality of signals for longer reads.
- Signal-to-noise ratio:
- Due to the factors mentioned above, the signal intensity for longer fragments is lower.
- This leads to a decreased signal-to-noise ratio for longer reads, making it more difficult to accurately determine the base calls at the end of a sequence.
Some students might suggest that the depletion of ddNTPs is responsible for the lower quality of longer reads. However, this is not correct. It's important to clarify that:
- The reaction mixture contains an excess of both dNTPs and ddNTPs.
- The concentrations of these nucleotides are not significantly depleted during the sequencing reaction.
- The relative concentrations of dNTPs and ddNTPs remain essentially constant throughout the process.
Q02¶
Points: 6
What is the purpose of adding adapters to DNA fragments in Illumina sequencing?
Solution
Adapters are short oligonucleotide sequences added to DNA fragments during Illumina sequencing library preparation.
Key purpose
Adapters contain sequences complementary to oligonucleotides on the Illumina flow cell surface. This allows DNA fragments to bind to the flow cell and form clusters.
They serve several other crucial purposes.
- Priming for sequencing: Adapters include primer binding sites for both forward and reverse sequencing reactions.
- Index/barcode sequences: Adapters often contain unique index sequences that allow for multiplexing—running multiple samples in a single sequencing lane.
- Bridge amplification: Adapter sequences facilitate bridge amplification, which generates clonal clusters of each DNA fragment on the flow cell surface.
- Sequencing initiation: The adapter sequences provide a known starting point for the sequencing reaction.
Q03¶
Points: 4
Compare and contrast the principles behind Sanger sequencing and Illumina sequencing. How does each method overcome the challenge of determining the order of nucleotides in a DNA strand? In your answer, consider the strengths and limitations of each approach.
Solution
Sanger Sequencing:
- Principle: Based on the selective incorporation of chain-terminating dideoxynucleotides (ddNTPs) by DNA polymerase during in vitro DNA replication.
- Method:
- Uses a single-stranded DNA template, a DNA primer, DNA polymerase, normal deoxynucleotides (dNTPs), and modified nucleotides (ddNTPs) that terminate DNA strand elongation.
- Resulting DNA fragments are separated by size using capillary electrophoresis.
- Determining sequence:
- Nucleotide order is determined by the length of DNA fragments produced when chain termination occurs.
- The fragments are separated by size, and the terminal ddNTP of each fragment indicates the nucleotide at that position.
- The sequence is read from the shortest to the longest fragment.
- Strengths:
- High accuracy for long reads (up to ~900 base pairs).
- Good for sequencing specific genes or DNA regions.
- Still considered the "gold standard" for validation of other sequencing methods.
- Limitations:
- Low throughput compared to next-generation methods.
- Higher cost per base compared to Illumina sequencing.
- Difficulty in sequencing low-complexity regions or regions with extreme GC content.
Illumina Sequencing:
- Principle: Uses sequencing by synthesis (SBS) technology, where fluorescently labeled nucleotides are detected as they are incorporated into growing DNA strands.
- Method:
- DNA is fragmented and adapters are ligated to both ends of the fragments.
- Fragments are amplified on a flow cell surface, creating clusters.
- Sequencing occurs in cycles, where a single labeled nucleotide is added, detected, and then the label is cleaved off before the next cycle.
- Determining sequence:
- Nucleotide order is determined by detecting the specific fluorescent signal emitted when each base is incorporated.
- Millions of clusters are sequenced simultaneously, with each cluster representing a single DNA fragment.
- The sequence of each cluster is built up one base at a time over multiple cycles.
- Strengths:
- Very high throughput, capable of sequencing millions of fragments simultaneously.
- Cost-effective for large-scale sequencing projects.
- Highly accurate due to the depth of coverage (each base is sequenced multiple times).
- Versatile, can be used for whole genome sequencing, transcriptomics, and more.
- Limitations:
- Shorter read lengths (typically 150-300 base pairs) compared to Sanger sequencing.
- Higher error rates in homopolymer regions.
- More complex data analysis required due to the large volume of data generated.
Crucial Differences:
- Scale and Throughput: Illumina sequencing is massively parallel, allowing for much higher throughput than Sanger sequencing.
- Read Length: Sanger produces longer individual reads compared to Illumina.
- Methodology: Sanger uses chain termination, while Illumina uses sequencing by synthesis.
- Application: Sanger is better for targeted sequencing of specific genes, while Illumina is preferred for whole genome or exome sequencing.
Subtle Differences:
- Sample Preparation: Illumina requires more complex library preparation, including adapter ligation and amplification.
- Error Profiles: Each method has different types of sequencing errors. Sanger tends to have higher error rates at the beginning and end of reads, while Illumina can struggle with GC-rich regions.
- Data Analysis: Illumina sequencing requires more sophisticated bioinformatics tools for data processing and analysis.
- Cost Structure: While Illumina is more cost-effective for large-scale projects, Sanger can be more economical for sequencing small numbers of samples or specific regions.
- Sensitivity: Illumina can detect low-frequency variants more easily due to its high depth of coverage, which is more challenging with Sanger sequencing.
Q04¶
Points: 5
In Sanger sequencing, the ratio of dideoxynucleotides (ddNTPs) to deoxynucleotides (dNTPs) is crucial for successful sequencing. Explain why this ratio is essential and predict what might happen if:
- The concentration of ddNTPs is too high;
- The concentration of ddNTPs is too low.
Solution
The optimal ratio of ddNTPs to dNTPs in Sanger sequencing is critical for generating a balanced distribution of fragment lengths. This ratio is essential for the following reasons:
- Fragment Distribution: The ratio ensures that chain termination occurs randomly at different positions along the template DNA, producing a set of fragments that differ in length by one nucleotide.
- Read Length: It allows for the generation of fragments covering the entire sequence of interest, typically up to 900-1000 base pairs.
If the concentration of ddNTPs is too high:
- Short Fragments: There would be an overabundance of short DNA fragments due to frequent early termination of DNA synthesis.
- Loss of Long Reads: Longer fragments would be underrepresented or absent, leading to incomplete sequence information for the latter part of the template.
- Reduced Overall Signal: The signal intensity would decrease rapidly along the length of the sequence, making it difficult to read bases accurately beyond the first 100-200 nucleotides.
If the concentration of ddNTPs is too low:
- Long Fragments: There would be an overabundance of long DNA fragments due to infrequent termination of DNA synthesis.
- Loss of Short Reads: Shorter fragments would be underrepresented, leading to poor sequence quality at the beginning of the read.
- Weak Signal for Short Fragments: The signal intensity for the first part of the sequence would be very low, making it difficult to accurately determine the initial bases.
Q05¶
Points: 4
Illumina sequencing uses "bridge amplification" to create clusters of identical DNA fragments. Describe how this process works and explain why it's necessary for the Illumina sequencing method. How does this compare to the amplification process in Sanger sequencing?
Solution
Bridge amplification is a crucial step in Illumina sequencing that creates clusters of identical DNA fragments on the surface of a flow cell. The process works as follows:
- Flow Cell Preparation: The flow cell surface is coated with two types of oligonucleotide adapters.
- DNA Fragment Attachment: Single-stranded DNA fragments with adapters at both ends are added to the flow cell. These fragments bind randomly to the complementary adapters on the surface.
- Bridge Formation: The free end of a bound DNA fragment bends over and hybridizes to a nearby complementary adapter on the surface, forming a "bridge."
- Amplification: DNA polymerase creates a complementary strand, forming a double-stranded bridge.
- Denaturation: The double-stranded bridge is denatured, resulting in two single-stranded copies of the molecule tethered to the flow cell surface.
- Repeated Cycles: Steps 3-5 are repeated multiple times, with each fragment amplifying into a distinct cluster.
- Reverse Strand Removal: One strand (usually the reverse strand) is cleaved and washed away, leaving clusters of single-stranded, identical DNA fragments.
Bridge amplification is necessary for Illumina sequencing for several reasons:
- Signal Amplification: It creates thousands of identical copies of each DNA fragment, significantly amplifying the signal for detection during sequencing.
- Clonal Clusters: Each cluster represents a single original DNA fragment, ensuring that the sequencing signal comes from identical molecules.
- Parallelization: Millions of clusters can be generated on a single flow cell, allowing for massive parallelization of sequencing reactions.
- Improved Accuracy: The clonal nature of each cluster helps to reduce sequencing errors by providing multiple identical copies for each base call.
- High Throughput: The dense arrangement of clusters on the flow cell surface enables high-throughput sequencing.
The amplification process in Illumina sequencing differs significantly from that used in Sanger sequencing:
- Method:
- Illumina: Uses solid-phase bridge amplification on a flow cell surface.
- Sanger: Typically uses solution-phase PCR or cloning in bacterial vectors.
- Scale:
- Illumina: Massively parallel, creating millions of clusters simultaneously.
- Sanger: Limited to amplifying one DNA fragment at a time.
- Product:
- Illumina: Generates clonal clusters of single-stranded DNA.
- Sanger: Produces a population of double-stranded DNA fragments.
- Locality:
- Illumina: Amplification occurs in a fixed location on the flow cell.
- Sanger: Amplification occurs in solution.
Q06¶
Points: 4
You are designing primers for a Sanger sequencing experiment. The region you want to sequence contains an important single nucleotide polymorphism (SNP) at position 90 from the start of your sequence of interest. Given what you know about the typical quality of Sanger sequencing reads at different positions, where would you design your sequencing primer to bind? Explain your reasoning.
Solution
Before designing the primer, it's crucial to understand the typical quality pattern of Sanger sequencing reads:
- Start of the read (first ~15-35 bases): Often low quality due to primer binding and initial reaction instability.
- Middle section (~35-700 bases): Highest quality, with accurate base calls.
- End of the read (beyond ~700-900 bases): Decreasing quality due to signal decay and increased noise.
Given that the SNP of interest is at position 90 from the start of the sequence, the ideal primer design would place this SNP within the high-quality middle section of the sequencing read. Specifically, design the primer to bind approximately 40-50 bases upstream of the SNP.
Rationale:
- This placement allows for the initial low-quality bases (first 15-35) to be read before reaching the SNP.
- It positions the SNP at around base 130-140 in the sequencing read (40-50 bases for primer + 90 bases to SNP).
- This location falls well within the high-quality middle section of a typical Sanger sequencing read.
Benefits of this design:
- Ensures high-quality base calls around the SNP position.
- Provides sufficient context before and after the SNP for accurate analysis.
- Allows for potential upstream sequence verification if needed.
Q07¶
Points: 2
You are conducting a Sanger sequencing experiment on a gene of interest isolated from a wild plant population. Upon examining the chromatogram, you notice that multiple peaks frequently overlap at several positions, making base calling difficult. This pattern persists even when you repeat the sequencing with newly designed primers. Further investigation reveals that the plant samples were collected from an area known for its high biodiversity and the presence of closely related species.
What is one possible molecular or genomic explanation for the overlapping peaks observed in your chromatogram? Describe how it could lead to the pattern in the sequencing results.
Clarification
In this scenario, we are working with plant samples collected from a wild population in an area known for high biodiversity. This means we're not dealing with lab-grown, genetically identical plants, but rather with plants that have natural genetic variation.
When we say we're sequencing "a gene of interest isolated from a wild plant population," we're typically working with DNA extracted from a single plant specimen. However, this single specimen can contain genetic material from multiple closely related organisms due to various biological phenomena.
Sanger sequencing typically starts with many copies of a specific DNA region (our "gene of interest"). These copies are created through PCR amplification of the extracted DNA. The sequencing reaction is then performed on this amplified DNA.
The chromatogram represents the sequencing results from all the DNA molecules present in your sample. If there are multiple versions of the gene present, they will all be sequenced simultaneously, resulting in overlapping peaks.
Solution
The observation of multiple overlapping peaks at several positions in a Sanger sequencing chromatogram, persisting even with newly designed primers, can be attributed to various molecular and genomic phenomena. Given the context of a wild plant population in an area of high biodiversity, several plausible explanations warrant consideration.
High Heterozygosity Due to Outcrossing
In outcrossing plant species, high levels of heterozygosity can accumulate, especially in regions with large, diverse populations.
- Molecular Mechanism
- Plants in the population cross-pollinate, leading to offspring with diverse allelic combinations.
- Over generations, multiple alleles for each gene can persist in the population.
- Individual plants may carry two or more distinct alleles for many genes.
- How it Causes Overlapping Peaks
- When sequencing a heterozygous individual, both alleles are amplified and sequenced.
- Differences between alleles result in double peaks at heterozygous positions.
- Supporting Evidence
- Consistent with a wild population in a biodiverse area.
- Might be particularly noticeable if the population has high genetic diversity.
Allopolyploidy
Allopolyploidy results from hybridization between two or more different species, followed by chromosome doubling.
- Mechanism
- Hybridization occurs between two related species (e.g., Species A: AA and Species B: BB).
- The resulting hybrid (AB) undergoes chromosome doubling to form an allopolyploid (AABB).
- The allopolyploid now contains two sets of similar but not identical genomes.
- How it Causes Overlapping Peaks
- Multiple similar copies of each gene are present in the genome.
- During PCR and sequencing, all copies are amplified and sequenced simultaneously.
- Where sequences differ between copies, multiple bases are incorporated, resulting in overlapping peaks.
- Supporting Evidence
- Consistent with the high biodiversity and presence of closely related species in the area.
- Explains the persistence of the pattern with new primers.
Q08¶
Points: 4
In Illumina sequencing, adapters are ligated to DNA fragments before sequencing.
- Explain the role of these adapters in the sequencing process.
- How might errors in adapter ligation affect the sequencing results and downstream data analysis?
- Describe how the design of these adapters helps prevent the formation of adapter dimers.
Solution
Adapters in Illumina sequencing play several crucial roles throughout the sequencing workflow:
- Enabling Bridge Amplification
- Adapters contain sequences complementary to oligonucleotides on the flow cell surface.
- This allows DNA fragments to bind to the flow cell and undergo bridge amplification.
- Priming for Sequencing
- Adapters include binding sites for sequencing primers.
- This enables the initiation of sequencing reactions for both forward and reverse reads.
- Index Sequences
- Adapters often contain index sequences (barcodes).
- These allow for multiplexing, where multiple samples can be sequenced in the same lane and later demultiplexed.
- Facilitating Paired-End Sequencing
- In paired-end sequencing, adapters at both ends of the fragment allow sequencing from both directions.
Errors in adapter ligation can have several negative effects on sequencing results and downstream analysis:
- Reduced Sequencing Efficiency
- Fragments without properly ligated adapters won't bind to the flow cell, reducing the overall yield.
- Chimeric Reads
- Improper ligation can lead to chimeric fragments where adapters join unrelated DNA fragments.
- This results in chimeric reads that can complicate assembly and alignment processes.
- Biased Representation
- Inefficient ligation can lead to under-representation of certain sequences, especially those with extreme GC content.
- This introduces bias in quantitative analyses like RNA-Seq or ChIP-Seq.
- Index Hopping
- In multiplexed samples, errors in index sequence ligation can lead to index hopping.
- This results in reads being assigned to the wrong sample during demultiplexing.
- Reduced Read Quality
- Partially ligated adapters can lead to poor quality reads, especially at the beginning or end of reads.
- Complications in Data Analysis
- Adapter contamination in reads can interfere with alignment, assembly, and variant calling.
- It necessitates more stringent quality control and adapter trimming steps in data preprocessing.
- False Positive Variant Calls
- Adapter sequences present in reads can be misinterpreted as real genomic sequence, leading to false positive variant calls.
Illumina adapters are designed with several features to minimize the formation of adapter dimers:
- Y-shaped Adapters
- Adapters are often designed with a Y-shape, where only a portion of the adapter is double-stranded.
- This reduces the likelihood of adapters ligating to each other.
- Sequence Design
- Adapter sequences are designed to minimize complementarity between different adapter molecules.
- This reduces the likelihood of base-pairing between adapters.
- PCR Suppression Effect
- Some adapter designs incorporate sequences that, when dimerized, form strong hairpin structures.
- These structures are poor substrates for PCR amplification, suppressing the amplification of adapter dimers.
Q09¶
Points: 5
Compare the structure of a typical surfactant to a phospholipid. How do these structural differences contribute to the surfactant's ability to lyse cells? Explain why increasing the concentration of surfactants in a lysis buffer might not always lead to better DNA yield. What potential problems could arise from using too much surfactant?
Solution
Structural Comparison: Surfactant vs. Phospholipid
Surfactants and phospholipids, while both amphipathic molecules, exhibit distinct structural characteristics that significantly influence their behavior in biological systems. A typical surfactant consists of a hydrophilic head group and a single hydrophobic tail. The head group can vary in its chemical nature, being ionic (either anionic or cationic), nonionic, or zwitterionic. The hydrophobic portion is usually a single tail, often composed of a hydrocarbon chain. Common examples of surfactants include sodium dodecyl sulfate (SDS), Triton X-100, and CTAB.
In contrast, phospholipids possess a more complex structure. They are composed of a hydrophilic head group and two hydrophobic tails. The head group typically contains a phosphate group and may include other molecules, such as choline in the case of phosphatidylcholine. The hydrophobic portion consists of two tails, usually fatty acid chains. Examples of phospholipids include phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine.
The key structural differences between surfactants and phospholipids lie in several aspects. Firstly, the number of hydrophobic tails differs, with surfactants typically having one and phospholipids having two. Secondly, the head group composition of surfactants is generally simpler compared to the more complex phosphate-containing heads of phospholipids. Thirdly, surfactants are generally smaller molecules than phospholipids in terms of molecular weight. Lastly, the overall geometry differs, with surfactants often having a conical shape, while phospholipids tend to be more cylindrical.
Structural Differences and Cell Lysis Ability
The structural disparities between surfactants and phospholipids significantly contribute to the surfactant's superior ability to lyse cells. The single tail and conical shape of surfactants allow them to insert themselves more easily between the phospholipids in the cell membrane. This insertion causes membrane curvature and eventually leads to the formation of mixed micelles containing both surfactants and membrane lipids, effectively disrupting the membrane structure.
Surfactants generally have a lower Critical Micelle Concentration (CMC) than phospholipids. At concentrations above the CMC, surfactants form micelles more readily, which enhances their ability to solubilize membrane components. The simpler structure of surfactants also allows them to interact more easily with membrane proteins, potentially denaturing them and further destabilizing the membrane.
The smaller size of surfactants enables them to diffuse and equilibrate across the membrane more quickly than phospholipids. Additionally, the variety of head groups available in surfactants (ionic, nonionic, zwitterionic) allows for the selection of surfactants that can optimally interact with different types of cell membranes, making them versatile tools for cell lysis in various applications.
Surfactant Concentration and DNA Yield
While surfactants are crucial for cell lysis, increasing their concentration in a lysis buffer might not always lead to better DNA yield. This counterintuitive phenomenon can be attributed to several factors.
Excessive surfactant concentrations can lead to extensive protein denaturation. While some protein denaturation is necessary for DNA release, overly denatured DNA-binding proteins can potentially lead to DNA degradation.
At high concentrations, surfactants can interact directly with DNA, potentially causing structural changes or precipitation, which can reduce yield. Above the CMC, additional surfactants form micelles rather than contributing to lysis, providing diminishing returns. Excessively high surfactant concentrations can also disrupt the careful balance of other buffer components, potentially affecting pH or ionic strength, which are crucial for optimal DNA extraction.
Furthermore, high surfactant concentrations can interfere with subsequent purification steps or analytical techniques, indirectly reducing the final DNA yield. The presence of excess surfactants may complicate downstream processes, leading to losses during purification or hindering accurate quantification of the extracted DNA.
Potential Problems from Excessive Surfactant Use
Using too much surfactant in cell lysis procedures can lead to several issues that may compromise the quality and quantity of the extracted DNA. Excessive surfactant can lead to DNA damage, including denaturation or fragmentation, especially in combination with other factors like heat or mechanical stress. Over-solubilization of cellular proteins can lead to higher protein contamination in the DNA extract, necessitating additional purification steps.
Residual surfactants can inhibit PCR and other enzymatic reactions, affecting downstream applications. High surfactant concentrations can interfere with alcohol-based DNA precipitation methods, reducing recovery. Excessive surfactant can also cause problematic foam formation during lysis and extraction procedures, making sample handling more difficult.
A02
Q01¶
Points: 7
You run FastQC on a set of sequencing reads and observe the following "Per base sequence quality" plot.
a) Describe what this plot is showing.
b) What does this pattern typically indicate about the sequencing run?
c) How might this affect downstream analyses if left uncorrected?

Solution
a
The "Per base sequence quality" plot from FastQC visually represents how the sequencing quality varies across each base position in the reads. On the horizontal axis, it displays base positions ranging from 1 to 239 bp, corresponding to the sequencing reads' length. The vertical axis shows the Phred quality scores from 0 to 40, indicating the confidence level in identifying each base; higher scores reflect greater confidence and lower error probabilities.
A box plot summarizes the distribution of quality scores from all reads at each base position along the X-axis. The central line within each box represents the median quality score, while the top and bottom edges of the box indicate the 25th and 75th percentiles, encompassing the middle 50% of the data. The whiskers extend to show the range of scores outside this interquartile range, highlighting the variability in quality at each position.
The plot's background is color-coded to facilitate quick assessment of quality levels: green indicates high-quality scores above 28, orange signifies reasonable quality scores between 20 and 28, and red denotes poor quality scores below 20. In this plot, the quality scores are consistently high and within the green zone from positions 1 to 150 bp, indicating strong confidence in base calling in this region. Beyond position 150 bp, there is a noticeable decline in quality. From positions 150 to 200 bp, the scores decrease but generally remain within the green zone. However, between positions 200 and 239 bp, there is a sharp decline, with quality scores dropping into the orange and even red zones. Additionally, the spread of the box plots widens towards the end of the reads, reflecting increased variability and uncertainty in the base calls in these later positions.
b
The observed pattern—where high-quality scores at the beginning of the reads progressively decline towards the end—is characteristic of data generated by high-throughput sequencing platforms like Illumina. This pattern indicates that while the initial portion of the sequencing run produced high-quality data, the latter part is compromised by decreased accuracy and increased variability, a common phenomenon but significant for data interpretation. This trend suggests that several factors contribute to a reduction in base-calling accuracy as the sequencing reaction proceeds. One key factor is signal decay; over successive cycles, the fluorescence signals used to identify nucleotides become weaker and less distinguishable, making accurate base identification more challenging. Issues with phasing and pre-phasing can also arise, where some DNA strands fall out of sync during the sequencing process, leading to cumulative errors in base calling. Instrument calibration or maintenance issues also impact the sequencing efficiency and accuracy over time.
c
If the decline in sequencing quality towards the end of the reads is not addressed, it can affect downstream analyses in several ways. In alignment tasks, low-quality bases at the ends of reads may lead to incorrect mapping to the reference genome, resulting in mismatches or gaps that reduce overall alignment accuracy and confidence. This misalignment can propagate errors into variant calling, where sequencing errors might be misinterpreted as genetic variants, leading to false positives, or genuine variants might be overlooked due to the masking effect of poor-quality data, resulting in false negatives.
In de novo assembly projects, low-quality regions can fragment contigs or cause misassemblies, as the assembly algorithms struggle to accurately piece together sequences with high error rates. For quantitative analyses like gene expression, including erroneous bases can lead to inaccurate conclusions about gene expression levels or community composition.
Moreover, retaining low-quality data increases computational demands and storage requirements without contributing valuable information, potentially slowing down analysis pipelines and complicating data management. To mitigate these issues, it is crucial to perform quality control measures such as trimming low-quality bases from the ends of reads using tools like Trimmomatic or Cutadapt. By removing or correcting poor-quality data, researchers can improve the reliability and accuracy of downstream analyses, ensuring that conclusions drawn from the sequencing data are based on high-quality, trustworthy information.
Q02¶
Points: 7
In a FastQC report, you notice a high percentage of overrepresented sequences that match known Illumina adapters.
a) What step in data preprocessing should you perform to address this issue?
b) How might the presence of adapter sequences affect genome assembly if not removed?
Solution
a
When you notice a high percentage of overrepresented sequences in a FastQC report that match known Illumina adapters, the appropriate step in data preprocessing is to perform adapter trimming. This involves using specialized software tools like Trimmomatic, Cutadapt, or Trim Galore to scan your sequencing reads and remove any adapter sequences present at the ends. By trimming these adapters, you ensure that downstream analyses—such as genome assembly or alignment—are based solely on genuine genomic data, enhancing the accuracy and reliability of your results.
b
If adapter sequences are not removed before genome assembly, their presence can adversely affect the assembly process in several ways. Adapter sequences can create erroneous overlaps between reads, leading the assembler to incorrectly join unrelated genomic fragments. This can result in misassembled contigs or scaffolds and the formation of chimeric contigs, where non-adjacent genomic regions are incorrectly assembled together due to shared adapter contamination.
Additionally, the presence of adapters can reduce the overall quality of the assembly by inflating error rates and decreasing assembly metrics like N50 values, leading to a fragmented and less accurate genome assembly.
Adapter contamination can also skew read depth and coverage analyses, affecting downstream interpretations such as gene expression levels or variant calling.
Furthermore, including adapter sequences increases the complexity of the dataset, leading to longer processing times and higher computational resource usage during assembly.
Q03¶
Points: 7
You observe in your "Per sequence GC content" plot in your FastQC report a bimodal distribution.
a) What does a bimodal distribution in this plot typically suggest?
b) List a possible reason for this observation.
c) How would you further investigate the cause of this pattern?
Solution
a
A bimodal distribution in the "Per sequence GC content" plot typically suggests that your sequencing data contains two distinct groups of sequences with different GC content percentages. This often indicates that there is more than one source of DNA in your sample. In other words, instead of all your reads having a similar GC content (which would result in a unimodal, bell-shaped distribution), the presence of two peaks suggests heterogeneity in the GC content of the sequences.
b) Possible Reason for the Observation
A possible reason for observing a bimodal distribution in the GC content plot is contamination with DNA from another organism that has a significantly different GC content than your target organism. This contamination could occur during sample collection, preparation, or sequencing.
Alternatively, the presence of technical sequences such as adapters, primers, or other synthetic constructs with distinct GC content can also contribute to the formation of a second peak in the distribution.
c) Investigating the Cause of the Bimodal Pattern
To further investigate the cause of this bimodal pattern, you can take the following steps:
- Sequence Alignment and BLAST Search:
- Extract Reads: Separate a subset of reads from each peak of the GC content distribution.
- BLAST Analysis: Perform BLAST searches of these reads against nucleotide databases to identify their potential origins.
- Interpret Results: Determine if the reads from the unexpected peak align to genomes of other organisms, which would indicate contamination.
- Mapping to Reference Genome:
- Alignment: Map all reads to your target organism's reference genome using alignment tools like BWA or Bowtie2.
- Assess Mapping Rates: Check if the reads from the second GC peak fail to map to the reference genome, suggesting they originate from a different organism.
- Taxonomic Classification:
- Use Metagenomic Tools: Employ tools like Kraken, Centrifuge, or Kaiju to classify the taxonomic origin of the reads.
- Quantify Contamination: Determine the proportion of reads assigned to organisms other than your target, providing evidence of contamination.
- Check for Technical Sequences:
- Screen for Adapters and Primers: Use software like FastQC or Cutadapt to identify and quantify any remaining adapter or primer sequences.
- Trim if Necessary: Remove any technical sequences and reassess the GC content distribution to see if the bimodal pattern persists.
- Review Laboratory Procedures:
- Sample Handling: Re-examine the sample preparation steps to identify any potential sources of contamination.
- Reagents and Equipment: Ensure that all reagents and equipment were free from contaminants and that proper aseptic techniques were followed.
- Consult Metadata and Experimental Design:
- Mixed Samples: Confirm whether the sample was supposed to contain multiple organisms (e.g., in metagenomic studies).
- Experimental Controls: Review any controls included in the experiment to rule out technical artifacts.
Q04¶
Points: 7
Explain the concept of \(k\)-mers in the context of genome assembly. How does the choice of \(k\)-mer size affect the assembly process? Provide examples of potential issues with very small and very large \(k\)-mer sizes.
Solution
In genome assembly, \(k\)-mers are all the possible substrings of length \(k\) that can be extracted from sequencing reads. For example, if you have a read sequence AGCTG, the 3-mers (where \(k\) = 3) would be AGC, GCT, and CTG.
\(K\)-mers are fundamental in assembly algorithms, particularly those using de Bruijn graphs, where nodes represent \(k\)-mers, and edges represent overlaps between \(k\)-mers.
By analyzing how these \(k\)-mers overlap, assemblers can reconstruct the original genome sequence.
The size of \(k\) in \(k\)-mers significantly impacts the assembly process:
- Smaller \(k\)-mers are more likely to find overlaps between reads because they require fewer matching bases. This increases sensitivity, helping to connect reads in regions with low coverage or sequencing errors. Larger \(k\)-mers are more specific, reducing the chance of erroneous overlaps but requiring higher-quality data.
- Genomes often contain repetitive sequences. Smaller \(k\)-mers may not distinguish between these repeats because the \(k\)-mers are too short to capture unique sequences, leading to ambiguities in the assembly. Larger \(k\)-mers can span unique regions within repeats, aiding in correctly assembling these areas.
- In de Bruijn graph assemblies, smaller \(k\)-mers result in graphs with more connections (edges), which can make the graph more complex and computationally intensive to resolve. Larger \(k\)-mers simplify the graph by reducing the number of connections but may fragment the graph if coverage is insufficient.
Potential Issues with Very Small and Very Large \(k\)-mer Sizes
- Very Small \(k\)-mer Sizes (e.g., k ≤ 15):
- High Ambiguity in Repeats: Short \(k\)-mers are common across the genome, especially in repetitive regions. This leads to multiple possible paths in the de Bruijn graph, making it challenging to determine the correct assembly path and potentially causing misassemblies.
- Error Propagation: Sequencing errors can introduce incorrect \(k\)-mers that overlap with many reads, creating false connections in the graph and complicating error correction.
- Computational Load: The increased number of connections can make the graph more complex and harder to resolve, increasing computational time and memory usage.
- Very Large \(k\)-mer Sizes (e.g., k ≥ 127):
- Fragmented Assemblies: Longer \(k\)-mers require contiguous sequences without errors to match perfectly. In regions with sequencing errors or low coverage, the necessary \(k\)-mers may be missing, leading to breaks in the assembly and resulting in fragmented contigs.
- Sensitivity to Sequencing Errors: A single nucleotide error can render a large \(k\)-mer unusable because the entire \(k\)-mer must match exactly. This sensitivity can significantly reduce the number of usable \(k\)-mers, especially in datasets with higher error rates.
- Reduced Overlap Detection: With longer \(k\)-mers, the likelihood of finding overlaps between reads decreases, which can hinder the assembly of regions where reads do not overlap sufficiently.
- Increased Memory Usage: Storing and handling a vast number of large \(k\)-mers can require more computational resources, potentially exceeding the capacity of standard computing environments.
Some modern assembly algorithms employ multiple \(k\)-mer sizes or adaptive \(k\)-mer strategies to leverage the advantages of different \(k\)-mer lengths. By carefully selecting and possibly combining different \(k\)-mer sizes, assemblers aim to produce the most accurate and contiguous genome assemblies possible given the data quality and computational resources available.
Q05¶
Points: 6
Describe the greedy algorithm approach to genome assembly:
a) Outline the basic steps of the algorithm.
b) What are the advantages and disadvantages of this approach?
c) Give an example scenario where the greedy algorithm might fail to produce the correct assembly.
Solution
a
The greedy algorithm is one of the earliest methods used for genome assembly and operates on a simple principle: at each step, it makes the locally optimal choice with the hope of finding a global optimum. In the context of genome assembly, the algorithm builds the genome sequence by iteratively merging reads based on the best available overlaps. Here are the basic steps:
- Compute All Pairwise Overlaps
- Calculate the overlaps between every possible pair of sequencing reads.
- Assign scores to overlaps based on their length and quality (longer overlaps with fewer errors receive higher scores).
- Select the Best Overlapping Pair
- Find the pair of reads with the highest-scoring overlap.
- This pair represents the most significant immediate opportunity for extension.
- Merge the Selected Pair
- Align the two reads at the overlapping region.
- If there are mismatches, resolve them (often by selecting the most common nucleotide or using quality scores).
- Combine the two reads into a single sequence (contig).
- Update the Read Set
- Exclude the original reads from the dataset.
- Treat the newly formed contig as a read for subsequent iterations.
- Repeat the Process
- Go back to step 2 and repeat the selection and merging process with the updated set of reads and contigs.
- Continue until no further overlaps exceed a predefined threshold, or all reads have been assembled into contigs.
- Output the Assembled Genome
- The remaining contigs represent the assembled genome sequences.
- Additional steps like scaffolding or gap filling may follow to improve the assembly.
b
Advantages:
- Simplicity and Ease of Implementation The algorithm is straightforward and does not require complex data structures. It is easy to understand and implement, making it accessible for educational purposes or small projects.
- Speed on Small Datasets For datasets with a small number of reads, the greedy algorithm can be relatively fast since it requires fewer computations.
- Immediate Results Since the algorithm makes the best local choice at each step, it quickly extends contigs, providing rapid preliminary assemblies.
- Minimal Computational Resources for Small Data Does not demand significant memory or processing power when dealing with limited datasets.
Disadvantages:
- Suboptimal Global Assembly The algorithm's local optimization does not guarantee a globally optimal solution. Early decisions may prevent better assemblies later on, leading to fragmented or incorrect genomes.
- Poor Handling of Repetitive Sequences Repeats in the genome can cause the algorithm to misassemble reads because it cannot distinguish between identical sequences from different locations. This leads to collapsed repeats or chimeric contigs.
- Sensitivity to Sequencing Errors Sequencing errors can mislead the algorithm into making incorrect merges or missing true overlaps. The algorithm lacks robust error correction mechanisms.
- Computational Inefficiency with Large Datasets Calculating all pairwise overlaps becomes computationally intensive as the number of reads increases. Does not scale well with high-throughput sequencing data common in modern genomics.
-
Lack of Quality Consideration Often, the algorithm does not adequately consider the quality scores of bases, which can result in assemblies that include erroneous sequences.
-
No Mechanism for Resolving Conflicts When multiple overlaps of similar length are found, the algorithm may arbitrarily choose one, potentially leading to incorrect assemblies.
c
Imagine you are assembling the genome of a plant species known to have a high content of repetitive DNA sequences, such as transposable elements, which can be several kilobases long and occur thousands of times throughout the genome.
- Reads originating from different copies of the repeat are nearly identical.
- The greedy algorithm, prioritizing the longest overlaps, cannot distinguish between overlaps from unique regions and those from repeats.
- The algorithm merges reads from different repeat copies, collapsing them into a single contig.
- This results in a reduced representation of the genome size and loss of repeat copy number information.
- Merging reads from different genomic locations creates contigs that do not exist in the actual genome.
- The flanking unique sequences of different repeats get erroneously joined.
- The algorithm may encounter conflicting overlaps of similar lengths and arbitrarily choose one, potentially breaking contigs or preventing the correct assembly of unique regions adjacent to repeats.
Q06¶
Points: 6
You are given the following set of reads from a DNA sequencing experiment:
{ATGGCTA, GGCTAAC, CTAACGT, AACGTAG, CGTAGCT, TAGCTAA, GCTAACG, TAACGTA, ACGTAGT}.
a) Using the greedy algorithm, show the steps to assemble these reads into contig(s). Assume a minimum overlap of 3 bases. For each step, clearly state which reads you are merging and the resulting sequence. If there are multiple possibilities with the same overlap length, explain your choice.
Clarification
By saying "Assume a minimum overlap of 3", this means you cannot make a merge if the overlap is less than 3. So if you get to a point where you have two sequences and they only have an overlap of 1, you cannot merge them.
If I didn't specify a minimum overlap, you still take the highest overlap each time but you don't have a minimum overlap requirement to merge. If the highest overlap was 2, you can still make that merge. If there is no overlap, you cannot concatenate.
Since this was confusing, I will accept any valid greedy assembly.
b) What is the final assembled sequence?
c) Is this assembly unique? Why or why not?
Clarification
"Unique assembly" in the context of DNA sequence assembly refers to a situation where there is only one possible way to combine the given reads (DNA fragments) to reconstruct the original sequence. In other words, if there are multiple valid ways to combine these reads then the assembly is not unique.
d) Identify a potential problem with this assembly that might not reflect the true original sequence. Explain your reasoning.
Solution
When assembling the given set of reads using the greedy algorithm with a minimum overlap of three bases, the process involves iteratively finding the pair of sequences with the maximum overlap and merging them until no further merges are possible. The reads provided are:
ATGGCTAGGCTAACCTAACGTAACGTAGCGTAGCTTAGCTAAGCTAACGTAACGTAACGTAGT
a
Step 1: Identify All Possible Overlaps
First, we compute all pairwise overlaps of at least three bases between the reads. We look for overlaps where the suffix of one read matches the prefix of another read. Here are the significant overlaps identified:
- Read 1 (ATGGCTA) overlaps with Read 2 (GGCTAAC) by five bases (
GGCTA). - Read 2 (GGCTAAC) overlaps with Read 7 (GCTAACG) by six bases (
GCTAAC). - Read 7 (GCTAACG) overlaps with Read 3 (CTAACGT) by six bases (
CTAACG). - Read 3 (CTAACGT) overlaps with Read 8 (TAACGTA) by six bases (
TAACGT). - Read 8 (TAACGTA) overlaps with Read 4 (AACGTAG) by six bases (
AACGTA). - Read 4 (AACGTAG) overlaps with Read 9 (ACGTAGT) by six bases (
ACGTAG). - Read 5 (CGTAGCT) overlaps with Read 6 (TAGCTAA) by five bases (
TAGCT).
Step 2: Merge Reads Based on Maximum Overlaps
We begin merging reads starting with the pairs that have the maximum overlap length.
Merge 1: Read 2 and Read 7
- Overlap: Six bases (
GCTAAC). - Merged Sequence:
GGCTAACG. - Reads Remaining: Reads 1, Merged Read (
GGCTAACG), Reads 3–6, 8, 9.
Merge 2: Merged Read and Read 3
- Overlap: Six bases (
CTAACG). - Merged Sequence:
GGCTAACGT. - Reads Remaining: Reads 1, Merged Read (
GGCTAACGT), Reads 4–6, 8, 9.
Merge 3: Merged Read and Read 8
- Overlap: Six bases (
TAACGT). - Merged Sequence:
GGCTAACGTA. - Reads Remaining: Reads 1, Merged Read (
GGCTAACGTA), Reads 4–6, 9.
Merge 4: Merged Read and Read 4
- Overlap: Six bases (
AACGTA). - Merged Sequence:
GGCTAACGTAG. - Reads Remaining: Reads 1, Merged Read (
GGCTAACGTAG), Reads 5, 6, 9.
Merge 5: Merged Read and Read 9
- Overlap: Six bases (
ACGTAG). - Merged Sequence:
GGCTAACGTAGT. - Reads Remaining: Reads 1, Merged Read (
GGCTAACGTAGT), Reads 5, 6.
Merge 6: Read 5 and Read 6
- Overlap: Five bases (
TAGCT). - Merged Sequence:
CGTAGCTAA. - Reads Remaining: Reads 1, Merged Read (
GGCTAACGTAGT), Merged Read (CGTAGCTAA).
Merge 7: Read 1 and Merged Read (GGCTAACGTAGT)
- Overlap: Five bases (
GGCTA). - Merged Sequence:
ATGGCTAACGTAGT. - Reads Remaining: Merged Read (
ATGGCTAACGTAGT), Merged Read (CGTAGCTAA).
b
After these merges, no further significant overlaps are found between the remaining sequences. The final assembly consists of two contigs:
- Contig 1:
ATGGCTAACGTAGT - Contig 2:
CGTAGCTAA
c
This assembly is not unique. The reason is that during the assembly process, multiple overlaps of the same length were available, and choices made at each step could lead to different assembly paths. For example, when merging reads with an overlap of six bases, there were several options, and selecting a different pair could have resulted in a different contig sequence. The greedy algorithm does not consider all possible combinations or the global optimal assembly, so the final result depends on the order in which overlaps are considered and merged.
d
A potential issue with this assembly is that it results in two separate contigs instead of a single contiguous sequence, which may not reflect the true original genome sequence. The inability to merge the two contigs suggests that there might be missing reads or insufficient overlap information to bridge the gap between them. Additionally, the greedy algorithm's nature means it makes local optimal choices without considering the overall assembly, potentially leading to misassemblies or fragmented genomes.
Specifically, the reads CGTAGCT and TAGCTAA were merged to form Contig 2, but this contig could not be connected to Contig 1. It's possible that there was an overlap between Contig 2 and an internal region of Contig 1 that was overlooked due to the algorithm's limitations or because the required overlap length was not met. This fragmentation could result in missing genetic information or misrepresentation of genomic structures, which is critical when accurate genome assembly is necessary for downstream analyses.
Q07¶
Points: 6
You are given the following set of reads from a DNA sequencing experiment:
@read1
ATGCGTAC
+
IIIIIIII
@read2
CGTACGTA
+
IIIIIFFF
@read3
CGTACATA
+
FFIIIIFF
@read4
TACGTAGT
+
FFHHIIII
a) Construct a de Bruijn graph for these reads using \(k\) = 5, where \(k\) is the edge length. Draw the graph, clearly labeling nodes and edges.
b) Identify and explain any features in your graph that might complicate genome assembly, such as bubbles/bulge, tips, or cycles.
c) Propose a possible original sequence that could have generated these reads. If multiple possibilities exist, explain why.
d) How would increasing \(k\)edge to 6 change the structure of the graph? Discuss both potential benefits and drawbacks of this change.
Note
In your de Bruijn graph, represent the frequency of each edge by labeling it with a number.
Solution
a
To construct a de Bruijn graph using the provided reads and a \(k\)-mer size of 5, we begin by extracting all possible 5-mers from the reads. In this context, the nodes of the graph will be all possible (\(k\)-1)-mers, which are 4-mers, and the edges will represent the 5-mers connecting these nodes.
For each read, we extract the 5-mers and determine the prefix and suffix 4-mers (nodes). Now we identify all unique nodes and edges:
ATGC,TGCG,GCGT,CGTA,GTAC,TACG,ACGT,TACA,ACAT,CATA,GTAG,TAGT.
flowchart LR
A((ATGC)) -->|1| B((TGCG))
B -->|1| C((GCGT))
C -->|1| D((CGTA))
D -->|1| E((GTAG))
E -->|1| F((TAGT))
D -->|3| G((GTAC))
G -->|1| H((TACG))
H -->|2| I((ACGT))
I -->|2| D
G -->|1| J((TACA))
J -->|1| K((ACAT))
K -->|1| L((CATA))b
The de Bruijn graph constructed exhibits several features that can complicate genome assembly:
Branches (Forks):
- At Node
CGTA: There are two outgoing edges leading toGTACandGTAG, creating ambiguity in the assembly path. - At Node
GTAC: There are two outgoing edges toTACGandTACA, further increasing complexity.
Cycles:
- Cycle Involving
CGTAandACGT: The pathCGTA→GTAC→TACG→ACGT→CGTAforms a loop. Cycles can cause assemblers to get stuck or repeat sections erroneously.
Bubbles/Bulges:
The presence of multiple paths between the same nodes can create bubbles in the graph, representing sequencing errors or genuine variations. These features introduce uncertainty in determining the correct path through the graph, making it challenging for assembly algorithms to reconstruct the original sequence accurately.
c
In order to determine possible contigs, we try various paths from the ATGC tip to either TAGT or CATA.
ATGCGTAGTATGCGTACATAATGCGTACGTAGTATGCGTACGTACATAATGCGTACGTACGTAGTATGCGTACGTACGTACATA
These contigs represent the main paths through the graph that don't involve repeating significant portions of the sequence. The circular path is included because it represents a potential repetitive element in the genome. It's worth noting that there could be variations of these contigs depending on how many times you traverse the circular path. However, without additional information about the expected genome structure or read coverage, it's difficult to determine how many times this repeat should be included in the final assembly.
d
Since we are already using a \(k\)-mer size of 5, increasing the \(k\)-mer size would mean moving to \(k\) = 6. With longer \(k\)-mers, there are fewer possible \(k\)-mers extracted from the reads, leading to a simpler graph.
The use of longer \(k\)-mers in genome assembly offers several potential benefits. Firstly, it can lead to improved resolution of repeats, as longer \(k\)-mers are better able to span repetitive regions that shorter \(k\)-mers might struggle to distinguish. This can reduce ambiguity in the assembly graph and potentially resolve branching caused by short repeats, resulting in a more linear graph structure. Additionally, longer \(k\)-mers can simplify the overall graph structure by reducing the number of nodes and edges, making the graph easier to navigate and assemble. This simplification often results in fewer cycles and bubbles caused by errors or repetitive \(k\)-mers. Furthermore, longer \(k\)-mers enhance specificity, as they are less likely to occur by chance, which increases confidence in the overlaps between reads.
However, the use of longer \(k\)-mers also comes with potential drawbacks. One significant issue is the increased fragmentation due to sequencing errors. Since longer \(k\)-mers are more sensitive to errors, a single base error can invalidate an entire \(k\)-mer, potentially breaking the path in the graph and leading to disconnected components and gaps in the assembly. Another drawback is the reduced coverage of \(k\)-mers. Longer \(k\)-mers naturally occur less frequently in the dataset, which can result in insufficient coverage to form connections between nodes. This may lead to the loss of low-abundance sequences and the potential exclusion of rare variants. Lastly, longer \(k\)-mers can make it more difficult to assemble low-complexity regions. Areas with low complexity or high GC content might not produce enough valid longer \(k\)-mers, potentially resulting in assembly gaps.
Q08¶
Points: 6
You are working on assembling a bacterial genome. After initial quality control and assembly, you notice that your assembly is highly fragmented with a low N50 value.
a) List three possible reasons for this poor assembly.
b) For each reason, suggest a strategy to improve the assembly.
c) How would you validate the quality of your improved assembly?
Solution
a
When assembling a bacterial genome, encountering a highly fragmented assembly with a low N50 value can be indicative of several underlying issues. One possible reason for this poor assembly is insufficient sequencing coverage. Low sequencing depth means there aren't enough overlapping reads to reconstruct the genome contiguously, leading to gaps and fragmented contigs. Without adequate coverage, the assembler cannot confidently piece together the genome, resulting in a low N50 value, which reflects the fragmentation of the assembly.
Another potential cause is the presence of high repetitive content in the genome. Bacterial genomes often contain repetitive elements such as transposons, insertion sequences, or ribosomal RNA operons. These repetitive sequences can confuse assembly algorithms because reads from different parts of the genome may appear identical. The assembler may incorrectly merge these reads or fail to assemble them altogether, leading to fragmented assemblies and decreased N50 values.
A third reason could be poor-quality sequencing data. High error rates, uneven quality across reads, or residual adapter sequences can hinder the correct alignment and overlap of reads during assembly. Sequencing errors introduce incorrect bases, making it difficult for the assembler to find true overlaps between reads. Adapter contamination and low-quality bases at the ends of reads can also prevent proper merging, resulting in a fragmented assembly.
b
To address these issues and improve the assembly, you can employ several strategies. If insufficient sequencing coverage is the problem, increasing the sequencing depth is essential. This can be achieved by performing additional sequencing runs to generate more data. Before doing so, it's helpful to assess your current coverage using computational tools that estimate coverage levels and determine how much additional data is needed. Ensuring that your library preparation methods minimize biases can also help achieve more even coverage across the genome, which is crucial for a successful assembly.
In the case of high repetitive content, utilizing long-read sequencing technologies and advanced assembly techniques can make a significant difference. Long-read platforms like PacBio or Oxford Nanopore Technologies produce reads that are long enough to span repetitive regions, helping to resolve ambiguities that short reads cannot. Implementing a hybrid assembly approach, where you combine short-read data (which is typically more accurate) with long-read data (which provides greater contiguity), can leverage the strengths of both technologies. Additionally, creating mate-pair libraries with larger insert sizes can help bridge gaps caused by repeats, connecting contigs that short-insert libraries cannot.
If poor-quality sequencing data is contributing to the fragmented assembly, improving data quality through stringent preprocessing is crucial. Start by assessing the quality of your reads using tools like FastQC, which can identify issues such as low-quality scores or adapter contamination. Trimming low-quality bases and removing adapter sequences using software like Trimmomatic or Cutadapt can enhance the overall quality of your dataset. Applying error correction algorithms can also rectify sequencing errors before assembly, increasing the likelihood of correct read overlaps. If data quality issues persist despite these measures, it may be necessary to resequence your samples using optimized protocols to obtain higher-quality reads.
c
Once you've implemented strategies to improve the assembly, validating the quality of the new assembly is an essential next step. Begin by evaluating assembly metrics such as N50 and L50 values. An increase in N50 and a decrease in the number of contigs indicate improved contiguity of the assembly. Comparing the total length of your assembly to the expected genome size for your bacterial species can also provide insight into its completeness.
Mapping the original sequencing reads back to the improved assembly is another critical validation step. Using alignment tools like BWA or Bowtie2, you can assess how well the reads align to the assembly. Uniform coverage across the genome suggests that the assembly accurately represents the sequencing data. Analyzing mismatch and insertion-deletion (indel) rates during alignment can help identify any regions with potential errors.
Employing tools like BUSCO (Benchmarking Universal Single-Copy Orthologs) can evaluate the completeness of your assembly by checking for the presence of conserved genes expected in your bacterial genome. A high percentage of complete BUSCO genes indicates that your assembly captures most of the essential genomic content. If a closely related reference genome is available, aligning your assembly to it using software like MUMmer can reveal structural variations or misassemblies, providing further validation.
Annotating the genome using tools such as Prokka can help identify genes, ribosomal RNA, transfer RNA, and other genomic features. Ensuring that essential metabolic pathways and housekeeping genes are present supports the functional completeness of your assembly. It's also important to check for contamination by using taxonomic classification tools like Kraken or Kaiju. These tools can detect and quantify any sequences that may have originated from other organisms, allowing you to exclude contaminant contigs from your assembly.
Analyzing repetitive regions with specialized software can assess how well these elements have been assembled. Verifying that repetitive sequences are accurately represented without fragmentation or misassembly is crucial, especially if your bacterial genome contains a high amount of repeats. Laboratory validation methods, such as PCR amplification of specific genomic regions followed by Sanger sequencing, can provide nucleotide-level confirmation of your assembly's accuracy.
Finally, conducting a comprehensive assessment using tools like QUAST (Quality Assessment Tool for Genome Assemblies) can generate detailed reports on various assembly metrics. Evaluating statistics such as GC content distribution, gene density, and \(k\)-mer frequencies can offer additional insights into the quality of your assembly.
Q09¶
Points: 8
Design a step-by-step workflow for preprocessing raw Illumina sequencing data and performing a de novo genome assembly. For each step, briefly explain its purpose and mention one commonly used bioinformatics tool that could be used to perform that step.
Solution
To preprocess raw Illumina sequencing data and perform a de novo genome assembly, you can follow a structured workflow that ensures each step enhances the quality and accuracy of the final assembly. The process begins with the quality assessment of the raw sequencing data, which is essential for identifying any potential issues that could affect downstream analyses. Tools like FastQC are commonly used for this purpose. FastQC generates detailed reports on various quality metrics, including per-base sequence quality, GC content, sequence length distribution, and the presence of overrepresented sequences or adapter contamination. By thoroughly examining these reports, you can detect problems such as low-quality reads or technical artifacts early in the process.
Following the quality assessment, the next crucial step is quality trimming and adapter removal. This step aims to eliminate low-quality bases from the ends of the reads and remove any residual adapter sequences that might have been introduced during the library preparation. Removing these unwanted sequences improves the overall quality of the data and prevents them from interfering with the assembly process. Trimmomatic is a widely used tool for this purpose. It can perform both quality trimming based on Phred quality scores and adapter removal using provided adapter sequence files. By specifying parameters tailored to your dataset, Trimmomatic efficiently cleans the reads, resulting in a dataset composed of high-quality sequences ready for assembly.
After trimming, it's beneficial to perform error correction on the cleaned reads to reduce the impact of sequencing errors, which can hinder the assembly process by creating false overlaps or introducing incorrect sequences. Error correction algorithms analyze the reads to identify and correct probable errors, enhancing the overall accuracy of the data. BayesHammer, part of the SPAdes assembler suite, is an effective tool for error correction of Illumina sequencing data. It employs a Bayesian approach to distinguish between sequencing errors and true polymorphisms, correcting errors while preserving genuine genetic variation.
With high-quality, error-corrected reads, the next step involves removing potential contaminant sequences. Contamination can originate from various sources, such as microbial DNA in the environment or other samples processed concurrently. Identifying and eliminating these contaminant reads ensures that the assembly reflects only the genome of the target organism. Kraken is a taxonomic classification tool that can efficiently assign reads to known organisms by matching \(k\)-mers to a database of reference sequences. By running Kraken on your dataset, you can detect and remove reads that map to non-target organisms, resulting in a purified dataset for assembly.
Now, you are ready to perform the de novo genome assembly. This process reconstructs the genome without the guidance of a reference sequence by assembling the reads based on their overlaps. SPAdes is a commonly used assembler for Illumina data, particularly well-suited for small genomes like those of bacteria. SPAdes uses a de Bruijn graph approach to assemble genomes from short reads, handling various read lengths and paired-end data effectively. By inputting your cleaned and corrected reads into SPAdes, the assembler constructs contigs and scaffolds that represent the genome sequence.
Once the assembly is complete, it's important to assess the quality of the assembled genome to ensure it meets the necessary standards for your research. Evaluating assembly metrics provides insights into the contiguity, completeness, and accuracy of the assembly. QUAST (Quality Assessment Tool for Genome Assemblies) is a tool designed for this purpose. QUAST generates comprehensive reports that include metrics like N50 (a statistic that indicates the contig length at which 50% of the genome is contained in contigs of that length or longer), total assembly length, number of contigs, GC content, and potential misassemblies. If a reference genome is available, QUAST can also compare your assembly against it to identify structural variations or errors.
Finally, to add functional context to your assembly, you should perform genome annotation. This step involves identifying genes, coding sequences, rRNAs, tRNAs, and other genomic features within the assembled genome, providing valuable insights into the organism's biology. Prokka is an automated annotation tool that streamlines this process for bacterial genomes. It integrates various databases and prediction tools to annotate genes quickly and accurately, producing files compatible with genome browsers and further analyses.
Programming+¶
These problems are not required and will not impact your BIOSC 1540 grade. The instructor will assess these separately to validate correctness without an assigned grade. Thus, you may work on these problems individually or in a team-based setting and "due" by the end of the semester. Happy coding!
Acceptable languages: Python v3.10+, Rust v1.80+, Mojo v24.4+
Files: FASTQ
Rewards
Engaging with these optional programming problems offers several valuable academic and professional growth opportunities.
- Consistent engagement with these Programming+ problems will allow me to write more detailed, compelling recommendation letters highlighting your computational skills. These personalized letters can significantly boost your applications for future academic programs, internships, or job opportunities.
- If there is enough interest, optional Friday recitations will be provided. This will give you individualized attention to accelerate learning and actionable feedback on your code and problem-solving approaches.
- Exceptional solutions may be featured on our course website with the students' permission. This is a way for us to recognize and appreciate your hard work and dedication to these problems.
Note
These problems would be similar to ones given in a major-only version of the class. Although, there would be more relevant instructions during class and would be given more than a week to complete.
P1¶
Write a script that does the following:
- Reads the FASTQ file.
- Calculates and saves the mean quality score for each read as a CSV. Your score should subtract the 33 from each ASCII encoding.
- Makes the "Per base sequence quality" as produced by FastQC.
Example output format:
P2¶
Implement a function that takes a DNA sequence and a \(k\)-mer size as input, and returns a dictionary of unique \(k\)-mer counts. Then use this function to:
- Count all 3-mers in the sequences from any FASTQ file.
- Print the top 5 most frequent 3-mers and their counts.
- Explain how this information might be useful in genome assembly.
P3¶
Implement a simple de Bruijn graph construction algorithm:
- Write a function that takes a list of reads and a \(k\)-mer size (for edges) as input.
- The function should return a dictionary representing the de Bruijn graph, where keys are (\(k-1\))-mers and values are lists of possible next bases.
- Use your function to construct a de Bruijn graph for the reads in a FASTQ file with k=4.
- Print the node with the highest out-degree (i.e., the (\(k-1\))-mer with the most possible next bases).
- Identify any bubbles in the graph (nodes with multiple outgoing edges that later reconverge).
If you can implement a function to traverse this graph and generate a possible assembly, explain your approach and show the result.
A03
Q01¶
Explain the difference between structural and functional annotation in gene prediction. Why are both important in genomics research?
Solution
Structural Annotation refers to the identification and mapping of the physical components of the genome. This includes delineating the locations of genes, exons, introns, promoters, and other regulatory elements. The primary objective of structural annotation is to construct a comprehensive framework that outlines the architecture of the genome. Techniques such as sequence alignment, ab initio prediction algorithms, and comparative genomics are commonly employed to accurately predict gene structures. For instance, structural annotation would determine where a gene begins and ends, the arrangement of its coding sequences, and the presence of any non-coding regions within the gene.
On the other hand, Functional Annotation delves into assigning biological roles and characteristics to the identified genomic elements. Once the structural components are mapped, functional annotation seeks to elucidate the roles of these genes and regulatory regions in cellular processes, pathways, and overall organismal biology. This involves predicting gene functions based on sequence similarity to known genes, identifying protein domains, inferring metabolic pathways, and associating genes with phenotypic traits or diseases. Tools such as gene ontology (GO) databases, pathway analysis software, and protein interaction networks are integral to functional annotation efforts.
The distinction between the two lies in their focus: structural annotation is concerned with "where" genes and elements are located within the genome, while functional annotation addresses "what" these genes and elements do biologically.
Both types of annotation are indispensable in genomics research for several reasons:
- Comprehensive Genome Understanding: Structural annotation provides the necessary map of the genome, serving as a foundation upon which functional insights are built. Without knowing the precise locations and structures of genes, it would be challenging to investigate their functions effectively.
- Facilitating Downstream Analyses: Functional annotation enables researchers to interpret the biological significance of genetic variations, identify potential targets for therapeutic intervention, and understand the molecular mechanisms underlying various traits and diseases.
- Enhancing Comparative Genomics: By combining structural and functional annotations, scientists can perform comparative analyses across different species, revealing evolutionary conservation and divergence in gene structure and function.
- Supporting Personalized Medicine: Accurate annotations are crucial for identifying genetic variants that may influence an individual's response to drugs or susceptibility to diseases, thereby advancing personalized healthcare strategies.
Q02¶
Compare and contrast global and local sequence alignment. Provide example(s) of when each would be more appropriate to use.
Solution
In the study of bioinformatics and computational biology, sequence alignment is a critical tool used to identify regions of similarity between biological sequences, which can indicate functional, structural, or evolutionary relationships. There are two primary types of sequence alignment: global and local. Understanding the distinctions between these two methods is essential for selecting the appropriate approach based on the specific objectives of a genomic analysis.
Global alignment involves aligning two sequences from end to end, encompassing their entire lengths. This method attempts to optimize the alignment across the whole sequence, ensuring that every nucleotide or amino acid is included in the comparison. Global alignment is particularly effective when the sequences being compared are of similar length and are expected to be largely similar across their entire span.
One of the most widely used algorithms for global alignment is the Needleman-Wunsch algorithm, which employs dynamic programming to find the optimal alignment by scoring matches, mismatches, and gaps. The algorithm constructs a matrix to evaluate all possible alignments and determines the one with the highest score, representing the best overall alignment between the two sequences.
Global alignment is most suitable when comparing orthologous genes—genes in different species that originated from a common ancestral gene and retain similar functions. For instance, aligning the entire coding sequences of the hemoglobin gene from humans and mice can provide insights into evolutionary conservation and functional similarities. Since these genes are expected to be similar in length and structure, a global alignment ensures that the entire gene sequences are compared comprehensively.
In contrast, local alignment focuses on identifying the most similar subsequences within two larger sequences. Instead of attempting to align the entire length of both sequences, local alignment seeks regions of high similarity, which may be significantly shorter than the full length of the sequences. This approach is advantageous when the sequences being compared contain conserved domains or motifs amidst regions of divergence.
The Smith-Waterman algorithm is the standard method for performing local alignments. Like its global counterpart, it uses dynamic programming but differs in that it allows for the identification of optimal local matches by resetting the scoring matrix when negative scores are encountered. This enables the algorithm to locate the best matching regions without being penalized by dissimilar regions elsewhere in the sequences.
Local alignment is particularly useful when comparing sequences that may contain functional domains or motifs within otherwise divergent regions. For example, when aligning a protein sequence to a large genomic DNA sequence to identify potential coding regions, local alignment can pinpoint specific exons or functional domains without requiring the entire genomic sequence to match the protein. Another common application is in the identification of conserved motifs within regulatory regions of genes across different species, where only certain segments are expected to be conserved.
Q03¶
Describe the role of gap penalties in sequence alignment algorithms. How do different types of gap penalties (e.g., linear vs. affine) affect alignment results?
Solution
Gap penalties are scoring mechanisms that penalize the introduction of gaps—insertions or deletions—during the alignment process. These penalties are essential for balancing the alignment to reflect biological realities, such as insertions or deletions that occur during evolution or genetic variation. Understanding the role and types of gap penalties is fundamental to achieving biologically meaningful and accurate alignments.
Role of Gap Penalties in Sequence Alignment
When aligning two sequences, gaps are introduced to account for insertions or deletions that allow for a better overall alignment of matching regions. However, without appropriate penalties, algorithms might introduce excessive gaps, leading to unrealistic alignments. Gap penalties serve to:
- Discourage Excessive Gaps: By assigning a cost to each gap introduced, the algorithm is incentivized to minimize the number of gaps, promoting alignments that require fewer insertions or deletions.
- Reflect Biological Reality: Different types of gaps (e.g., single nucleotide insertions versus large indels) have varying biological implications. Gap penalties help model these differences by assigning appropriate costs.
- Optimize Alignment Scores: Gap penalties are integrated into the scoring system of alignment algorithms to ensure that the overall alignment score accurately reflects the quality of the alignment, balancing matches, mismatches, and gaps.
Types of Gap Penalties
Gap penalties can be broadly categorized into two types: linear and affine. Each type influences the alignment results differently based on how gaps are penalized.
Linear Gap Penalties
Linear gap penalties assign a fixed cost for each gap position, regardless of the length of the gap. The total penalty for a gap of length \(k\) is simply \(\text{gap penalty} \times k\).
-
Mathematical Representation: If the penalty per gap is \(g\), then a gap of length \(k\) incurs a penalty of \(g \times k\).
-
Equal Penalization: Each additional gap position is equally penalized, making longer gaps increasingly costly.
- Alignment Characteristics: This approach tends to favor multiple shorter gaps over fewer longer ones because the penalty accumulates linearly with gap length.
Linear gap penalties are suitable for alignments where insertions and deletions are expected to occur sporadically and independently throughout the sequence, without forming extended indels.
Affine Gap Penalties
Affine gap penalties differentiate between the initiation and extension of gaps by assigning separate penalties for gap opening and gap extension. Typically, the penalty is structured as:
where \(k\) is the length of the gap.
If the gap opening penalty is \(g_o\) and the gap extension penalty is \(g_e\), then a gap of length \(k\) incurs a penalty of \(g_o + g_e \times (k - 1)\).
- Differentiated Penalization: Opening a new gap incurs a higher penalty, while extending an existing gap is less costly.
- Alignment Characteristics: This approach favors the creation of longer, contiguous gaps over multiple shorter gaps because the relative cost of extending a gap is lower than opening a new one.
Affine gap penalties are ideal for alignments where insertions or deletions tend to occur in blocks or segments, reflecting scenarios such as exon-intron boundaries in genes or structural variations in genomes.
Comparative Effects on Alignment Results
The choice between linear and affine gap penalties significantly influences the resulting alignment:
| Aspect | Linear Gap Penalty | Affine Gap Penalty |
|---|---|---|
| Penalty Structure | Fixed cost per gap position | Higher cost for gap initiation, lower cost for extension |
| Tendency in Alignments | Favors multiple short gaps | Favors fewer, longer gaps |
| Biological Relevance | Suitable for random, independent indels | Suitable for grouped indels or structural variations |
| Algorithm Complexity | Simpler to implement and compute | More complex due to separate handling of gap openings and extensions |
| Use Cases | Aligning sequences with scattered mutations | Aligning sequences with potential large insertions/deletions or domain rearrangements |
Illustrative Example
Consider aligning two sequences where one sequence has a segment that is entirely inserted compared to the other. Using a linear gap penalty, the algorithm might introduce multiple single-nucleotide gaps to accommodate the insertion, incurring a penalty for each gap. In contrast, with an affine gap penalty, the algorithm is more likely to introduce a single contiguous gap covering the entire inserted segment, resulting in a lower total penalty and a more biologically plausible alignment.
Q04¶
Given the following DNA sequences:
ATGCACTAGCTAATGCTACGTA
Perform the following tasks:
a) Perform a global alignment using the Needleman-Wunsch algorithm. Use a scoring system of +1 for matches, -1 for mismatches, and -2 for gaps. Show your work, including the scoring matrix and traceback path.
Solution
The Needleman-Wunsch algorithm is a dynamic programming method used for global sequence alignment. It aligns two sequences from start to end, optimizing the alignment score based on a predefined scoring system. Below, we'll perform a global alignment of the given DNA sequences using the Needleman-Wunsch algorithm with the specified scoring system:
- Match: +1
- Mismatch: -1
- Gap: -2
Given Sequences
- Sequence 1 (S1):
ATGCACTAGCTA(Length: 12) - Sequence 2 (S2):
ATGCTACGTA(Length: 10)
For clarity, we index the sequences starting from 1:
- S1: A1 T2 G3 C4 A5 C6 T7 A8 G9 C10 T11 A12
- S2: A1 T2 G3 C4 T5 A6 C7 G8 T9 A10
Step 1: Initialize the Scoring Matrix
The scoring matrix has dimensions (m+1) x (n+1), where m and n are the lengths of S1 and S2, respectively. The first row and first column are initialized based on gap penalties.
- Rows: Represent characters from
S1(including a leading gap). - Columns: Represent characters from
S2(including a leading gap).
Given:
- m = 12 (Length of S1)
- n = 10 (Length of S2)
Initialization Rules:
- First Cell (0,0): 0
- First Row (i=0): Each cell
(0,j)=(0,j-1)+ Gap Penalty - First Column (j=0): Each cell
(i,0)=(i-1,0)+ Gap Penalty
Step 2: Construct and Fill the Scoring Matrix
Below is the fully constructed scoring matrix. Each cell (i,j) represents the optimal score for aligning the first i characters of S₁ with the first j characters of S₂.
Scoring Matrix:
| - | A1 | T2 | G3 | C4 | T5 | A6 | C7 | G8 | T9 | A10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | -2 | -4 | -6 | -8 | -10 | -12 | -14 | -16 | -18 | -20 |
| A1 | -2 | 1 | -1 | -3 | -5 | -7 | -9 | -11 | -13 | -15 | -17 |
| T2 | -4 | -1 | 2 | 0 | -2 | -4 | -6 | -8 | -10 | -12 | -14 |
| G3 | -6 | -3 | 0 | 3 | 1 | -1 | -3 | -5 | -7 | -9 | -11 |
| C4 | -8 | -5 | -2 | 1 | 4 | 2 | 0 | -2 | -4 | -6 | -8 |
| A5 | -10 | -7 | -4 | -1 | 2 | 3 | 3 | 1 | -1 | -3 | -5 |
| C6 | -12 | -9 | -6 | -3 | 0 | 1 | 2 | 4 | 2 | 0 | -2 |
| T7 | -14 | -11 | -8 | -5 | -2 | 1 | 0 | 2 | 3 | 3 | 1 |
| A8 | -16 | -13 | -10 | -7 | -4 | -1 | 2 | 0 | 1 | 2 | 4 |
| G9 | -18 | -15 | -12 | -9 | -6 | -3 | 0 | 1 | 1 | 0 | 2 |
| C10 | -20 | -17 | -14 | -11 | -8 | -5 | -2 | 1 | 0 | 0 | 0 |
| T11 | -22 | -19 | -16 | -13 | -10 | -7 | -4 | -1 | 0 | 1 | -1 |
| A12 | -24 | -21 | -18 | -15 | -12 | -9 | -6 | -3 | -2 | -1 | 2 |
Step 3: Traceback to Determine the Optimal Alignment
Traceback Process:
- Start: Bottom-right cell
(A12, A10)with a score of 2. - Goal: Reach the top-left cell
(0,0).
At each step, determine the direction from which the current cell's score was derived:
- Diagonal (↖): Match/Mismatch
- Up (↑): Gap in S2
- Left (←): Gap in S1
Traceback Steps:
- Cell (A12, A10): Score = 2
- Diagonal (A11, T9): Score = 1
- Characters:
AvsA(Match) - Calculated:
1 + 1 = 2→ Equal to current score (2)
- Characters:
- Up (T₁₁, A10): Score = -1
- Calculated:
-1 + (-2) = -3→ Less than current score (2)
- Calculated:
- Left (A₁₂, T₉): Score = -1
- Calculated:
-1 + (-2) = -3→ Less than current score (2)
- Calculated:
- Alignment Action: Match (
Aaligned withA)
- Diagonal (A11, T9): Score = 1
- Cell (T11, T9): Score = 1
- Diagonal (C10, G8): Score = 0
- Characters:
TvsT(Match) - Calculated:
0 + 1 = 1→ Equal to current score (1)
- Characters:
- Up (C10, T9): Score = 0
- Calculated:
0 + (-2) = -2→ Less than current score (1)
- Calculated:
- Left (T11, G8): Score = 0
- Calculated:
0 + (-2) = -2→ Less than current score (1)
- Calculated:
- Chosen Move: Diagonal (From C10, G8)
- Alignment Action: Match (
Taligned withT)
- Diagonal (C10, G8): Score = 0
- Continue Traceback: Repeat the process for each preceding cell, choosing the move that leads to the current cell's score.
Due to the complexity and size of the matrix, we'll summarize the final alignment based on the matrix and traceback.
Step 4: Final Alignment
Based on the traceback, the optimal global alignment between S₁ and S₂ with a final score of 2 is as follows:
Alignment Details:
- Matches: Positions where characters are identical.
- Mismatches: Positions with different characters.
- Gaps: Indicated by '-' where a character is missing in one sequence.
b) Using the same sequences and scoring system as above, perform a local alignment using the Smith-Waterman algorithm. Show your work, including the scoring matrix and traceback path.
Solution
| - | A1 | T2 | G3 | C4 | T5 | A6 | C7 | G8 | T9 | A10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| A1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| T2 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| G3 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| C4 | 0 | 0 | 0 | 1 | 4 | 2 | 0 | 1 | 0 | 0 | 0 |
| A5 | 0 | 1 | 0 | 0 | 2 | 3 | 3 | 1 | 0 | 0 | 1 |
| C6 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 4 | 2 | 0 | 0 |
| T7 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 2 | 3 | 3 | 1 |
| A8 | 0 | 1 | 0 | 0 | 0 | 0 | 3 | 1 | 1 | 2 | 4 |
| G9 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 2 | 2 | 0 | 2 |
| C10 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 1 | 1 | 0 |
| T11 | 0 | 0 | 1 | 0 | 0 | 3 | 1 | 0 | 1 | 2 | 0 |
| A12 | 0 | 1 | 0 | 0 | 0 | 1 | 4 | 2 | 0 | 0 | 3 |
c) Compare your results from the global and local alignments. What differences do you notice? Discuss the biological implications of these differences.
Solution
In comparing the global alignments produced by the Needleman-Wunsch algorithm with the local alignments generated by the Smith-Waterman algorithm, several key differences emerge that have significant biological implications.
- Scope of Alignment:
- Global Alignment (Needleman-Wunsch): This approach attempts to align the entire length of both sequences. Both alignments cover the full sequences of S1 and S2, introducing gaps to accommodate differences. For instance, in the first global alignment: Gaps are inserted in S2 to align with S1's additional characters.
- Local Alignment (Smith-Waterman): This method focuses on finding the most similar subsequences within the larger sequences, without necessarily aligning the entire sequence. The local alignments provided highlight highly similar regions, such as:
- Introduction of Gaps:
- Global Alignment: Gaps are systematically introduced to ensure that every part of both sequences is aligned, which can sometimes result in multiple gaps or extensive shifting to accommodate overall sequence differences.
- Local Alignment: Gaps are introduced selectively to optimize the alignment of highly similar regions, often resulting in fewer and strategically placed gaps that do not affect unrelated regions of the sequences.
- Handling of Sequence Length Differences:
- Global Alignment: Requires that the entire length of both sequences be accounted for, which can be problematic when sequences vary significantly in length or contain non-homologous regions.
- Local Alignment: More flexible in handling sequences of different lengths by focusing only on the most relevant and similar segments, ignoring the non-matching regions.
Biological Implications of These Differences:
- Functional Insights:
- Global Alignment: Best suited for comparing sequences that are expected to be entirely homologous, such as orthologous genes across different species where the overall structure and function are conserved. This comprehensive alignment can reveal large-scale similarities and differences, which are crucial for understanding evolutionary relationships and functional conservation.
- Local Alignment: Ideal for identifying conserved domains, motifs, or functional regions within larger proteins or genes that may have diverged otherwise. For example, a protein may contain a highly conserved active site amid regions of variability; local alignment would effectively highlight such critical areas without being confounded by less relevant differences.
- Evolutionary Studies:
- Global Alignment: Facilitates the study of sequence evolution over entire genes or genomes, allowing researchers to detect patterns of conservation and divergence that inform phylogenetic analyses.
- Local Alignment: Enables the detection of evolutionary conserved elements that may be under selective pressure, even if the surrounding regions have undergone significant changes. This is particularly useful for identifying conserved regulatory elements or protein-binding sites.
- Practical Applications:
- Global Alignment: Useful in applications where complete sequence similarity is required, such as in genome assembly, where overlapping sequences must be aligned across their entire lengths.
- Local Alignment: Essential in database searches (e.g., BLAST) where researchers seek to find regions of similarity between a query sequence and a vast database of sequences, identifying potential functional or evolutionary relationships without the need for complete sequence alignment.
Q05¶
a) List and briefly describe three key challenges in prokaryotic gene annotation.
Solution
Prokaryotic gene annotation involves the identification and characterization of genes within prokaryotic genomes, such as those of bacteria and archaea. While significant progress has been made in sequencing prokaryotic genomes, accurately annotating these genes remains challenging. Below are three key challenges in prokaryotic gene annotation, each accompanied by a brief description.
Functional Annotation of Hypothetical Proteins
A substantial proportion of genes in prokaryotic genomes encode proteins with unknown functions, often labeled as "hypothetical proteins." These proteins lack characterized homologs in existing databases, making it difficult to predict their roles based solely on sequence information.
The presence of numerous hypothetical proteins hampers the comprehensive understanding of cellular processes and metabolic pathways within prokaryotes. Without functional annotation, it is challenging to elucidate the organism's physiology, ecological roles, and potential applications in biotechnology or medicine.
Challenges:
- Limited experimental data to support functional predictions.
- Reliance on computational methods that may produce ambiguous or inaccurate annotations.
- Difficulty in distinguishing between genuinely novel proteins and misannotated genes.
Accurate Gene Prediction in Compact and Overlapping Genomes
Prokaryotic genomes are typically compact, with genes densely packed and often overlapping or arranged in operons. Additionally, prokaryotes may possess short open reading frames (ORFs) that are difficult to distinguish from non-coding regions.
Accurate identification of gene boundaries is crucial for downstream analyses, such as understanding gene regulation and protein function. Overlapping genes and compact genome organization increase the complexity of gene prediction algorithms, potentially leading to missed genes or incorrect annotations.
Challenges:
- Differentiating between closely spaced or overlapping genes.
- Identifying small genes that may be overlooked by prediction tools.
- Accounting for alternative start codons and non-standard genetic codes prevalent in some prokaryotes.
Identification and Annotation of Regulatory Elements and Operon Structures
Prokaryotic genes are often organized into operons—clusters of genes transcribed as a single mRNA molecule and regulated collectively. Identifying the boundaries of operons and the associated regulatory elements, such as promoters and operators, is essential for understanding gene regulation.
Incomplete or inaccurate annotation of regulatory elements can obscure the mechanisms controlling gene expression, affecting interpretations of gene function and interactions. Proper annotation is vital for constructing regulatory networks and modeling cellular responses to environmental changes.
Challenges:
- Variability in promoter sequences and regulatory motifs across different prokaryotic species.
- Limited conservation of regulatory elements, complicating comparative genomics approaches.
- Difficulty in experimentally validating predicted regulatory regions due to the vast number of potential sites.
b) What role do ribosomal binding sites (RBS) play in prokaryotic gene prediction? How are they typically identified?
Solution
Ribosomal binding sites (RBS), also known as Shine-Dalgarno (SD) sequences in bacteria, are short nucleotide motifs located upstream of the start codon of mRNA transcripts. These sequences are crucial for the initiation of translation, the process by which ribosomes synthesize proteins based on the genetic information encoded in mRNA. The primary functions of RBS in prokaryotic gene prediction include.
The RBS interacts with the complementary sequence on the 16S rRNA component of the small ribosomal subunit. This interaction aligns the ribosome with the start codon, ensuring accurate initiation of translation. Effective ribosome binding is essential for efficient protein synthesis and proper gene expression.
In gene prediction algorithms, the presence of an RBS serves as a key indicator of a potential translation start site. By identifying RBS motifs, computational tools can more accurately locate the boundaries of open reading frames (ORFs), distinguishing true genes from non-coding regions.
Incorporating RBS identification into gene prediction pipelines improves the precision of gene models. It reduces false positives by ensuring that predicted genes possess the necessary regulatory elements for translation and increases confidence in the functional annotation of genes.
Identification of Ribosomal Binding Sites
Accurate identification of RBS is integral to effective gene prediction in prokaryotes. Several methodologies and computational strategies are employed to detect RBS motifs within genomic sequences:
The most common RBS motif in bacteria is the Shine-Dalgarno sequence, typically characterized by a consensus sequence such as AGGAGG.
Gene prediction tools scan genomic regions upstream of potential start codons (usually within 10-20 nucleotides) for matches to this consensus sequence.
High-scoring matches increase the likelihood that the downstream ORF is a genuine gene.
Instead of relying solely on exact consensus sequences, PSSMs account for variability and degeneracy within RBS motifs. By assigning scores to each nucleotide position based on its likelihood of occurrence in true RBS sequences, these matrices allow for more flexible and sensitive detection of ribosomal binding sites, accommodating natural variations across different prokaryotic species.
Advanced gene prediction algorithms utilize machine learning techniques to identify RBS motifs. By training models on known RBS sequences, these approaches can learn complex patterns and features associated with ribosome binding. Machine learning models, such as hidden Markov models (HMMs) or neural networks, can integrate multiple signals, including sequence context and secondary structure, to improve the accuracy of RBS detection.
Comparative analysis across related prokaryotic genomes can enhance RBS identification. Conserved RBS motifs in orthologous genes across different species provide additional evidence for the presence of functional ribosomal binding sites. This evolutionary perspective helps distinguish genuine RBS sequences from random motifs that might otherwise be misidentified.
The secondary structure of mRNA near the RBS can influence ribosome binding efficiency. Computational tools that predict RNA secondary structures can identify RBS regions that are accessible and not occluded by hairpins or other structural elements. Incorporating secondary structure information ensures that identified RBS motifs are not only sequence-compatible but also structurally favorable for ribosome interaction.
Integration into Gene Prediction Pipelines
Effective prokaryotic gene prediction pipelines integrate RBS identification with other genomic signals to enhance overall annotation quality. The typical workflow includes:
- ORF Identification: Initial detection of potential ORFs based on start and stop codons, gene length, and codon usage bias.
- RBS Detection: Scanning regions upstream of identified ORFs for RBS motifs using the aforementioned methods. The presence and quality of an RBS contribute to the confidence score of the predicted gene.
- Functional Annotation: Combining RBS information with other annotation data, such as gene conservation, functional domains, and expression data, to assign functional roles to predicted genes.
- Validation and Refinement: Experimental data, such as ribosome profiling or transcriptomics, may be used to validate and refine gene predictions, ensuring that identified RBS motifs correspond to actively translated genes.
c) Explain the importance of identifying regulatory elements in gene annotation. Give two examples of regulatory elements and their functions.
Solution
In the field of genomics, regulatory elements are crucial components that control the expression of genes. Accurate identification and annotation of these elements are essential for understanding how genes are turned on or off in different cellular contexts, developmental stages, or environmental conditions. This understanding is fundamental to deciphering the complex regulatory networks that govern biological processes, disease mechanisms, and phenotypic diversity.
Importance of Identifying Regulatory Elements in Gene Annotation
- Understanding Gene Expression Regulation: Regulatory elements serve as control points that determine when, where, and how much a gene is expressed. By identifying these elements, researchers can map the regulatory landscape of the genome, revealing how genes are coordinated in response to internal signals and external stimuli. This is essential for elucidating the mechanisms underlying cellular differentiation, development, and homeostasis.
- Deciphering Complex Regulatory Networks: Genes do not operate in isolation; they are part of intricate networks that interact with multiple regulatory elements. Identifying these elements enables the construction of comprehensive gene regulatory networks, which are vital for understanding systems biology and the integration of various signaling pathways.
- Facilitating Functional Genomics and Disease Research: Many genetic variations associated with diseases are located in regulatory regions rather than within protein-coding sequences. Accurate annotation of regulatory elements allows for the identification of disease-associated variants that may disrupt normal gene regulation, thereby contributing to the development of targeted therapies and personalized medicine approaches.
- Enhancing Comparative Genomics and Evolutionary Studies: Comparative analysis of regulatory elements across different species can provide insights into evolutionary conservation and divergence of gene regulation. This helps in identifying fundamental regulatory mechanisms that are preserved through evolution and those that have adapted to species-specific needs.
- Improving Gene Prediction Algorithms: Incorporating regulatory elements into gene annotation pipelines enhances the accuracy of gene prediction by providing additional evidence for gene boundaries and functional annotation. This reduces false positives and improves the reliability of genomic annotations.
Examples of Regulatory Elements and Their Functions
Promoters
Promoters are DNA sequences located immediately upstream of the transcription start site (TSS) of a gene. They serve as binding sites for RNA polymerase and various transcription factors, initiating the process of transcription. Promoters play a critical role in determining the basal level of gene expression and are essential for the proper initiation of transcription.
- Core Promoter Elements: These include the TATA box, located approximately 25-30 base pairs upstream of the TSS, which helps position the RNA polymerase II machinery. Other elements such as the Initiator (Inr) and Downstream Promoter Element (DPE) also contribute to transcription initiation.
- Regulatory Sequences: Proximal promoter elements, located within a few hundred base pairs of the TSS, bind specific transcription factors that modulate the efficiency and rate of transcription in response to cellular signals.
In the lac operon of Escherichia coli, the promoter region contains the -10 (Pribnow box) and -35 sequences, which are recognized by the sigma factor of RNA polymerase. Binding of RNA polymerase to these promoter regions initiates the transcription of genes involved in lactose metabolism when lactose is present in the environment.
Enhancers
Enhancers are regulatory DNA sequences that can be located upstream or downstream of the gene they regulate, sometimes even hundreds of kilobases away from the transcription start site. Unlike promoters, enhancers can function in an orientation-independent manner and can act over long distances to increase the transcriptional activity of target genes.
- Binding Sites for Transcription Factors: Enhancers contain multiple binding sites for specific transcription factors, which, when bound, facilitate the recruitment of the transcriptional machinery to the promoter region.
- Chromatin Looping: Enhancers often interact with promoters through the formation of chromatin loops, bringing the enhancer-bound transcription factors into close proximity with the promoter to enhance transcription.
The beta-globin locus control region (LCR) in humans is an example of an enhancer. The LCR contains multiple enhancer elements that interact with the promoters of the beta-globin genes to regulate their expression during different developmental stages. This ensures the proper production of hemoglobin subunits necessary for oxygen transport in red blood cells.
Q06¶
a) What is the significance of using multiple k-mer sizes in SPAdes? How does this strategy improve assembly quality?
Solution
In the field of genome assembly, k-mers—substrings of length k derived from sequencing reads—are fundamental building blocks used to reconstruct the original genomic sequence. SPAdes (St. Petersburg genome assembler) is a widely used assembler that employs a sophisticated strategy involving multiple k-mer sizes to enhance assembly quality. Understanding the significance of using multiple k-mer sizes in SPAdes and how this approach improves assembly outcomes is essential for appreciating its effectiveness in handling complex genomic data.
Significance of Using Multiple k-mer Sizes in SPAdes
- Capturing Diverse Read Lengths and Coverage:
- Variable Read Coverage: Sequencing data often exhibit regions with varying coverage depths due to biases in sequencing technologies. Smaller k-mers can capture high-coverage regions effectively, while larger k-mers are better suited for low-coverage areas.
- Handling Read Length Variability: Sequencing reads can vary in length and quality. Multiple k-mer sizes allow SPAdes to adapt to these variations, ensuring that both short and long read segments are accurately represented in the assembly process.
- Resolving Repetitive Regions:
- Complex Repeats: Genomes contain repetitive sequences of varying lengths. Smaller k-mers may fail to resolve long repeats, leading to fragmented assemblies, whereas larger k-mers can span these repeats, aiding in their proper assembly.
- Balancing Specificity and Sensitivity: Using multiple k-mer sizes enables SPAdes to balance the specificity of larger k-mers (which reduce ambiguity in repeat regions) with the sensitivity of smaller k-mers (which ensure connectivity across the genome).
- Enhancing Graph Connectivity and Accuracy:
- De Bruijn Graph Complexity: Genome assembly algorithms like SPAdes construct De Bruijn graphs based on k-mers. Varying k-mer sizes help in creating a more connected and accurate graph by providing multiple perspectives on the sequence data.
- Error Correction: Multiple k-mer sizes facilitate more robust error correction by cross-validating information across different k-mer scales, thereby reducing the impact of sequencing errors on the final assembly.
How Multiple k-mer Sizes Improve Assembly Quality
- Improved Contiguity and Continuity:
- Longer Contigs: Larger k-mers contribute to longer contig assembly by bridging gaps across repetitive or low-complexity regions, resulting in more contiguous and complete genomic assemblies.
- Scaffolding Efficiency: The integration of multiple k-mer sizes enhances scaffolding by providing overlapping information that links contigs into larger scaffolds, improving the overall continuity of the assembly.
- Enhanced Resolution of Structural Variations:
- Detecting Insertions and Deletions: Multiple k-mer sizes allow SPAdes to more accurately identify and assemble regions with structural variations, such as insertions and deletions, by leveraging different levels of sequence resolution.
- Accurate Representation of Complex Genomic Features: Complex genomic features, including tandem repeats and structural rearrangements, are better resolved through the combined use of small and large k-mers, leading to a more faithful representation of the genome.
- Reduction of Misassemblies and Errors:
- Error Minimization: The cross-validation provided by multiple k-mer sizes helps in distinguishing true genomic sequences from erroneous k-mers, thereby reducing the likelihood of misassemblies.
- Robustness Against Sequencing Errors: Sequencing errors can introduce incorrect k-mers that complicate the assembly process. Multiple k-mer sizes enhance the assembler's ability to identify and discard these erroneous k-mers, improving the overall accuracy of the assembly.
- Flexibility Across Diverse Genomic Contexts:
- Adaptability to Different Genome Sizes and Complexities: Genomic projects often involve a wide range of organisms with varying genome sizes and complexities. The use of multiple k-mer sizes in SPAdes provides the necessary flexibility to effectively assemble genomes across this diversity.
- Scalability to High-Throughput Data: As sequencing technologies generate increasingly large and complex datasets, the multi k-mer approach ensures that SPAdes can scale efficiently, maintaining high assembly quality even with massive amounts of data.
Illustrative Example
Consider the assembly of a bacterial genome with several repetitive regions interspersed between unique sequences:
- Single k-mer Assembly: Using a single small k-mer size (e.g., k=21) might result in fragmented assemblies due to the inability to span long repetitive regions, leading to breaks in contigs where repeats occur.
- Multiple k-mer Assembly (SPAdes): By employing multiple k-mer sizes (e.g., k=21, 33, 55), SPAdes can utilize smaller k-mers to assemble highly covered unique regions and larger k-mers to bridge across repetitive sequences. This results in longer contigs that more accurately reflect the true genomic structure, minimizing fragmentation and improving overall assembly continuity.
b) What are "bulges" and "tips" in the context of the SPAdes assembly graph? How does SPAdes handle these structures?
Solution
In genome assembly, particularly within the framework of De Bruijn graphs, the construction and simplification of these graphs are pivotal for accurately reconstructing the original genomic sequence from short sequencing reads. SPAdes (St. Petersburg genome assembler) is an advanced assembler that employs sophisticated strategies to manage and resolve various structural complexities within the assembly graph. Two such complexities are known as "bulges" and "tips." Understanding these structures and how SPAdes handles them is essential for appreciating the assembler's effectiveness in producing high-quality genome assemblies.
Understanding Bulges and Tips in Assembly Graphs
Bulges
Bulges are structural irregularities within a De Bruijn graph that represent alternative paths between two nodes (k-1)-mers. They typically arise due to:
- Sequencing Errors: Errors in sequencing reads can introduce incorrect k-mers that create diverging paths in the graph.
- Genetic Variations: Natural variations such as single nucleotide polymorphisms (SNPs) or small insertions/deletions (indels) can lead to alternative sequences that form separate branches in the graph.
- Low Coverage Regions: Areas with insufficient sequencing coverage may result in incomplete or fragmented paths, leading to bulges.
Characteristics of Bulges:
- Parallel Paths: Bulges consist of multiple paths that start and end at the same nodes, creating a "bubble-like" structure.
- Short Length: Typically, bulges are short in length, corresponding to minor variations or errors.
- Resolution Requirement: Accurate genome assembly necessitates resolving these bulges to determine the correct path that represents the true genomic sequence.
Tips
Tips are another form of irregularity in the assembly graph, characterized by dead-end paths—at the beginning or end—that branch off from the main graph. They commonly result from:
- Sequencing Artifacts: Errors such as misincorporated bases or chimeric reads can create spurious k-mers that lead to short, dead-end branches.
- Low Coverage or Unique Regions: Regions with very low coverage or unique sequences may not have enough supporting reads to form continuous paths, resulting in tips.
- Collapsed Repeats: Highly repetitive regions can sometimes cause the assembler to create tips if the repeats are not properly resolved.
Characteristics of Tips:
- Dead-Ends: Tips are short branches that do not reconnect with the main graph, terminating or starting abruptly.
- Short Length: They are generally short in length, often representing minor sequencing errors or genuine biological variations.
- Potential for Removal: Many tips are artifacts and can be safely removed to simplify the graph without losing significant genomic information.
How SPAdes Handles Bulges and Tips
SPAdes employs a multi-step process to detect and resolve bulges and tips within the assembly graph, enhancing the accuracy and contiguity of the final genome assembly. The strategies involve error correction, graph simplification, and careful selection of optimal paths.
Error Correction and Preprocessing
Before addressing bulges and tips, SPAdes performs error correction on the sequencing reads. This step reduces the occurrence of erroneous k-mers that could contribute to bulges and tips. Error correction involves:
- K-mer Counting: SPAdes counts the occurrences of each k-mer across all reads to identify and correct errors.
- Consensus Building: By leveraging multiple overlapping reads, the assembler can determine the most probable sequence, correcting errors that appear infrequently.
Bulge Resolution
To resolve bulges, SPAdes employs the following strategies:
- Bubble Popping Algorithm: SPAdes identifies bubbles (bulges) by locating parallel paths between the same pair of nodes. It then evaluates these paths based on criteria such as k-mer coverage, path length, and consistency with the overall graph structure.
- Coverage-Based Selection: The assembler typically selects the path with higher coverage as the true sequence, assuming that genuine genomic sequences are more frequently represented in the reads compared to error-induced variations.
- Path Consistency: SPAdes ensures that the selected path maintains continuity and does not introduce further inconsistencies in the graph.
- Local Graph Reconciliation: After selecting the optimal path, SPAdes merges the nodes and edges along this path, effectively removing the alternative, less-supported branches that constitute the bulge.
Tip Removal
Handling tips involves identifying and eliminating short, dead-end branches—either at the beginning or end—that are likely artifacts rather than genuine genomic sequences. SPAdes employs the following methods:
- Length Thresholding: SPAdes sets a predefined length threshold below which tips are considered too short to represent true genomic variations. Tips shorter than this threshold are pruned from the graph.
- Coverage Filtering: Tips with significantly lower coverage compared to the main graph are likely to be sequencing errors and are removed. This is based on the assumption that genuine genomic sequences are consistently covered across multiple reads.
- Isolation Criteria: Tips that do not reconnect with the main graph and have no supporting evidence from overlapping reads are identified as candidates for removal.
Multi-Kmer Approach Enhancement
SPAdes’s use of multiple k-mer sizes enhances its ability to handle bulges and tips effectively:
- Small k-mers: Smaller k-mers are more sensitive to variations and can help in detecting subtle differences that form bulges. They are also useful in resolving tips by providing detailed local information.
- Large k-mers: Larger k-mers offer greater specificity, aiding in distinguishing between genuine repetitive regions and spurious branches. They help in accurately resolving bulges by providing longer contiguous sequences for comparison.
By integrating information from multiple k-mer sizes, SPAdes can more robustly identify and correctly resolve bulges and tips, ensuring that the final assembly accurately reflects the true genomic sequence.
Iterative Assembly Refinement
SPAdes often employs an iterative assembly process where the assembly graph is progressively refined:
- Graph Simplification: After initial resolution of bulges and removal of tips, SPAdes further simplifies the graph by collapsing linear paths and merging highly connected nodes.
- Reassembly Checks: The assembler may revisit previously resolved regions to ensure that no new bulges or tips have emerged due to the changes in the graph structure.
- Consensus Validation: SPAdes validates the resolved graph against the original read data to confirm that the chosen paths are consistent with the sequencing evidence.
Illustrative Example
Consider the assembly of a bacterial genome with a region containing a repetitive sequence flanked by unique sequences:
- Initial Graph Construction: SPAdes constructs a De Bruijn graph using multiple k-mer sizes, capturing both small-scale variations and long repetitive regions.
- Bulge Detection: A bulge forms in the graph due to a small indel in some reads, creating an alternative path between two nodes. SPAdes identifies this bubble and assesses the coverage of each path.
- Path Selection: The assembler determines that the main path has higher coverage, indicative of the true genomic sequence, and removes the alternative path representing the indel.
- Tip Identification: A short dead-end branch appears due to a sequencing error introducing a spurious k-mer. SPAdes detects the tip based on its short length and low coverage and removes it from the graph.
- Graph Refinement: The assembler merges the nodes along the selected path and simplifies the graph, ensuring a contiguous and accurate representation of the genomic region.
- Final Assembly: The resolved and simplified graph is translated into a contiguous sequence (contig) that accurately reflects the original genome, free from artifacts caused by bulges and tips.
c) Explain the concept of "paired-end reads" and how SPAdes utilizes this information to improve assembly.
Solution
Paired-end reads refer to a sequencing approach where both ends of a DNA fragment are sequenced, producing two reads per fragment with a known approximate distance between them, commonly referred to as the insert size. Unlike single-end reads, which sequence only one end of a DNA fragment, paired-end sequencing provides two reads that are spatially linked, offering additional contextual information about the original DNA molecule.
- Dual Sequencing: Both ends of a DNA fragment are sequenced, yielding two reads per fragment.
- Insert Size Information: The approximate distance between the two reads (insert size) is known, facilitating the estimation of the fragment's length.
- Orientation: The reads are typically oriented toward each other, with each read facing the opposite direction.
SPAdes Utilization of Paired-End Reads
SPAdes employs a sophisticated assembly strategy that integrates paired-end read information to construct accurate and contiguous genome assemblies. The assembler's approach leverages the dual nature of paired-end reads to overcome challenges inherent in genome assembly, such as repetitive sequences and structural complexities.
Incorporation into De Bruijn Graph Construction
SPAdes utilizes the De Bruijn graph paradigm for genome assembly, where sequences are broken down into smaller subsequences called k-mers. The integration of paired-end reads into this framework occurs through the following mechanisms:
- Graph Connectivity: The known insert size and orientation of paired-end reads provide additional connectivity information within the De Bruijn graph. This helps in accurately linking k-mers that belong to the same genomic region but are separated by repetitive or ambiguous sequences.
- Error Correction: Paired-end information aids in identifying and correcting sequencing errors by cross-validating the linkage between k-mers, ensuring that the graph reflects the true genomic structure.
Resolving Repetitive Regions
One of the primary challenges in genome assembly is the presence of repetitive sequences, which can lead to ambiguities in the assembly graph. Paired-end reads contribute significantly to resolving these regions:
- Bridging Repeats: The insert size information allows SPAdes to determine the correct placement of repeats by ensuring that the paired reads map to distinct and non-overlapping regions, thereby disambiguating repeat copies.
- Path Selection: When multiple paths exist within the graph due to repeats, the paired-end linkage information guides SPAdes in selecting the path that maintains consistent insert sizes and orientations, leading to a more accurate assembly.
Enhancing Contig and Scaffold Construction
Paired-end reads facilitate the extension and scaffolding of contigs, which are continuous sequences assembled from overlapping reads:
- Contig Extension: The proximity information from paired-end reads enables SPAdes to extend contigs by aligning reads that span the ends of contigs, thereby increasing their length and reducing fragmentation.
- Scaffolding: By linking contigs based on paired-end read connections, SPAdes can order and orient contigs into larger scaffolds. This process not only extends the assembly but also provides a higher-level organization of the genome structure.
Facilitating Gap Filling and Error Correction
The known insert sizes and orientations of paired-end reads aid in identifying and filling gaps within the assembly:
- Gap Identification: Discrepancies between expected and observed insert sizes can signal the presence of gaps or misassemblies, prompting further investigation and correction.
- Error Correction: Paired-end information helps in detecting and rectifying misassemblies or incorrect base calls by providing corroborative evidence from both ends of the DNA fragment.
Mechanisms of SPAdes in Leveraging Paired-End Information
SPAdes employs several computational strategies to effectively utilize paired-end read data:
- Multiple K-mer Sizes: SPAdes operates with a range of k-mer sizes, allowing it to capture both short and long-range linkage information provided by paired-end reads. This multi k-mer approach enhances the assembler's ability to handle complex genomic regions.
- Iterative Assembly Process: SPAdes iteratively refines the assembly graph by progressively integrating paired-end information. This iterative approach ensures that the assembler can resolve ambiguities and improve assembly accuracy with each iteration.
- Graph Simplification and Resolution: SPAdes simplifies the De Bruijn graph by resolving bulges (alternative paths) and tips (dead-end paths) using paired-end linkage information. This leads to a cleaner and more accurate assembly graph, reducing the likelihood of misassemblies.
- Error Correction Integration: Paired-end reads assist in the error correction phase by providing consistency checks across the linked reads, ensuring that the corrected k-mers are supported by multiple read pairs.
Impact on Assembly Quality
The incorporation of paired-end reads into SPAdes' assembly pipeline yields several tangible improvements in assembly quality:
- Increased Contiguity: Paired-end information reduces fragmentation by enabling the assembler to bridge gaps and extend contigs, resulting in longer and more contiguous assemblies.
- Enhanced Accuracy: The dual-read linkage provides robust evidence for correct sequence placement, minimizing errors and misassemblies, particularly in repetitive or complex genomic regions.
- Better Resolution of Structural Variants: Paired-end reads facilitate the detection and accurate assembly of structural variants, contributing to a more comprehensive and accurate representation of the genome.
Illustrative Example
Consider the assembly of a bacterial genome containing a region with a 500 base-pair (bp) tandem repeat:
- Single-End Assembly Challenge: Using only single-end reads, the assembler might struggle to determine the exact number of repeat units due to identical sequences, leading to fragmented contigs or misassemblies.
- Paired-End Assembly Advantage: With paired-end reads where each read pair spans the repeat region, the known insert size allows SPAdes to accurately link the repeat units. The paired reads provide the necessary linkage information to resolve the exact number of repeats, resulting in a contiguous and accurate assembly of the repetitive region.
Q07¶
a) You are assembling a bacterial genome using SPAdes. The assembly results in several large contigs and many small contigs. What might this indicate about the genome or sequencing data, and what steps could you take to improve the assembly?
Solution
When assembling a bacterial genome using SPAdes, encountering an assembly characterized by several large contigs alongside numerous small contigs can provide valuable insights into both the genomic features of the organism and the quality of the sequencing data. Understanding the underlying causes of such assembly patterns is crucial for diagnosing potential issues and implementing effective strategies to enhance assembly quality. Below, we explore the possible implications of this assembly outcome and outline actionable steps to improve the assembly process.
Possible Implications of Large and Small Contigs
Genome Complexity and Repetitive Regions
- Repetitive Elements: Bacterial genomes may contain repetitive sequences, such as insertion sequences, transposons, or ribosomal RNA (rRNA) operons. These repeats can complicate assembly by causing ambiguity in read alignment, leading to breaks in the assembly graph and the formation of smaller contigs.
- Multiple Replicons: Some bacteria possess multiple replicons, including plasmids and secondary chromosomes. These additional genetic elements can result in separate large contigs if they are not fully integrated into the main assembly.
Sequencing Data Quality and Coverage
- Uneven Coverage: Variability in sequencing coverage across the genome can lead to regions with insufficient data for robust assembly. Low-coverage areas may fail to bridge gaps between contigs, resulting in fragmented assemblies with many small contigs.
- Sequencing Errors: High error rates, particularly in specific regions of the genome, can disrupt the continuity of the assembly by introducing false k-mers that fragment the assembly graph.
Library Preparation and Read Length
- Short Read Lengths: Shorter sequencing reads may struggle to span longer repetitive regions or structural variants, making it difficult for SPAdes to assemble contiguous sequences, thereby producing numerous small contigs.
- Library Insert Size: Inappropriate insert sizes during library preparation can affect the ability of SPAdes to link contigs effectively, especially in repetitive or complex genomic regions.
Contamination and Mixed Populations
- Contaminant DNA: Presence of contaminant DNA from other organisms can introduce additional contigs that do not correspond to the target bacterial genome, contributing to the abundance of small, unrelated contigs.
Strategies to Improve Assembly Quality
To address the issues indicated by the presence of large and small contigs, the following steps can be undertaken to enhance the assembly quality:
- Increase Sequencing Depth: Ensuring adequate coverage (typically >50× for bacterial genomes) can provide sufficient data to resolve complex regions and reduce the likelihood of fragmented assemblies.
- Uniform Coverage: Utilize sequencing platforms and protocols that promote uniform coverage across the genome to minimize regions with low data density.
- Incorporate Long-Read Sequencing: Integrating long-read technologies, such as PacBio or Oxford Nanopore, can bridge repetitive regions and structural variants, resulting in more contiguous assemblies. SPAdes supports hybrid assembly approaches that combine short and long reads.
- High-Quality Reads: Employ stringent quality control measures to filter out low-quality reads and reduce sequencing errors that can disrupt assembly continuity.
- Appropriate Insert Sizes: Ensure that library preparation protocols generate insert sizes compatible with SPAdes' multi k-mer approach. Proper insert size selection facilitates effective linking of contigs and improves scaffolding.
- Mate-Pair Libraries: In addition to paired-end reads, incorporating mate-pair libraries with larger insert sizes can provide long-range linkage information, aiding in the resolution of repetitive regions and enhancing assembly contiguity.
- Error Correction Tools: Utilize pre-assembly error correction tools, such as BayesHammer (integrated within SPAdes), to correct sequencing errors before assembly, thereby reducing the formation of false k-mers that fragment the assembly graph.
- Contamination Screening: Employ tools like DeconSeq or BlobTools to identify and remove contaminant sequences from the dataset, ensuring that the assembly focuses solely on the target bacterial genome.
- K-mer Size Selection: Experiment with different k-mer sizes within SPAdes to find the optimal balance between sensitivity and specificity. SPAdes’ multi k-mer approach typically uses a range of k-mer sizes, but customizing this range based on the specific genome characteristics can yield better results.
- Coverage Cutoffs: Fine-tune coverage cutoffs to exclude low-abundance k-mers that may represent sequencing errors or contaminants, thereby simplifying the assembly graph and reducing fragmentation.
- Scaffolding Software: After initial assembly with SPAdes, employ scaffolding tools like SSPACE or Pilon to further link contigs based on paired-end information, improving assembly contiguity.
- Gap Filling: Use gap-filling tools such as GapCloser to resolve small gaps between contigs, enhancing the overall completeness of the assembly.
- Reassembly: Perform iterative assemblies by progressively incorporating additional data or adjusting assembly parameters based on the initial results. This iterative approach can help resolve persistent fragmentation issues.
- Validation with Reference Genomes: If available, compare the assembly against closely related reference genomes to identify and correct misassemblies or unresolved regions.
- Combine Multiple Assemblers: Integrate results from different assemblers to leverage their unique strengths, potentially resulting in a more complete and accurate assembly.
- Use of Long and Short Reads Together: Combining short-read assemblies with long-read data in a hybrid assembly framework can maximize the advantages of both sequencing technologies, leading to higher-quality assemblies.
b) Describe how you would use the SPAdes assembly graph visualization tool Bandage to analyze the quality of your assembly. What features would you look for?
Solution
When analyzing the assembly graph in Bandage, several features and indicators are pivotal in assessing assembly quality:
- Large Connected Components: A high-quality assembly typically exhibits a single, large connected component, indicating that most contigs are interconnected without fragmentation.
- Multiple Components: The presence of multiple large connected components may suggest contamination, incomplete assembly, or the existence of multiple replicons (e.g., plasmids) within the genome.
- Long Contigs: The presence of several large contigs is generally favorable, indicating that the assembler successfully resolved extensive genomic regions.
- Numerous Small Contigs: An assembly characterized by many small contigs alongside large ones may indicate unresolved repetitive regions, low coverage areas, or sequencing errors leading to fragmentation.
- Graph Loops: Loops or cycles within the assembly graph can signify unresolved repeats or structural variations. Excessive looping may complicate the assembly, leading to misassemblies or incomplete contigs.
- Resolution Strategies: Identifying loops allows for targeted refinement, such as adjusting k-mer sizes or incorporating long-read data to bridge repetitive regions.
- Bulges: These are parallel paths between two nodes, often representing alternative sequences due to sequencing errors or genuine genetic variations (e.g., SNPs). Bulges can fragment the assembly and should be resolved to maintain contiguity.
- Tips: Short, dead-end branches in the graph typically result from sequencing errors, low coverage, or misassemblies. Tips usually represent minor artifacts and can often be pruned to simplify the assembly graph.
- Scaffolding: SPAdes attempts to link contigs into scaffolds using paired-end read information. In Bandage, scaffolds appear as interconnected sequences that reflect the spatial arrangement of contigs in the genome.
- Gaps and Linkers: Bandage can visualize gaps between contigs within scaffolds, often represented by stretches of unknown bases (e.g., Ns). The size and distribution of these gaps provide insights into assembly completeness and potential regions needing improvement.
- Abrupt Path Terminations: Unexpected breaks or sudden changes in the graph structure may indicate misassemblies, where incorrect overlaps or erroneous contig connections have occurred.
- Comparison with Reference Genomes: If a reference genome is available, overlaying it with the assembly graph can help identify discrepancies, misassemblies, or structural variations.
Upon identifying potential issues through Bandage visualization, several corrective actions can be undertaken to improve assembly quality:
- Adjust K-mer Sizes: Experiment with different k-mer sizes in SPAdes to better resolve repetitive regions or improve contiguity. Smaller k-mers can enhance connectivity, while larger k-mers may help span repeats.
- Coverage Cutoffs: Modify coverage thresholds to exclude low-coverage k-mers that may represent sequencing errors or contaminants, thereby simplifying the assembly graph.
- Long-Read Technologies: Integrate long-read sequencing data (e.g., PacBio or Oxford Nanopore) to bridge repetitive regions and improve assembly continuity. SPAdes supports hybrid assembly approaches that combine short and long reads.
- Mate-Pair Libraries: Utilize mate-pair or linked-read libraries with larger insert sizes to provide long-range linkage information, aiding in the resolution of complex genomic structures.
- Gap Filling: Employ gap-filling tools to resolve stretches of unknown bases within scaffolds, enhancing assembly completeness.
- Error Correction: Use polishing tools (e.g., Pilon) to correct residual sequencing errors, improving the accuracy of the assembled contigs.
- Filter Contaminants: Identify and remove contaminant sequences using tools like DeconSeq or BlobTools to ensure that the assembly represents the target genome exclusively.
- Reassemble with Refined Data: After implementing corrective measures, perform an iterative assembly process to progressively enhance assembly quality based on insights gained from Bandage visualization.
Q08¶
a) You are given the following bacterial DNA sequence:
5' ATGCATGCTAGCTAATGCCCGGGTAACCCATGA 3'
Identify all possible ORFs in this sequence (consider both strands).
How would you determine which ORF is most likely to be a real gene?
Solution
To identify Open Reading Frames (ORFs) within a given DNA sequence, it is essential to analyze both strands of the DNA double helix. ORFs are continuous stretches of nucleotides that begin with a start codon (typically ATG) and terminate with a stop codon (TAA, TAG, or TGA). Identifying ORFs is a fundamental step in gene prediction, as it highlights potential protein-coding regions within the genome.
Given the bacterial DNA sequence:
we will systematically identify all possible ORFs on both the forward strand and the reverse complement (reverse) strand. Subsequently, we will discuss criteria to determine which ORF is most likely to represent a genuine gene.
Identifying ORFs on the Forward Strand
The forward strand is the sequence provided:
Step-by-Step Identification:
- Scanning for Start Codons (ATG): The sequence is scanned in the 5' to 3' direction for the ATG start codon.
- Determining Reading Frames: There are three possible reading frames on each strand, corresponding to the three possible positions where translation can start.
- Identifying ORFs: Once a start codon is found, the sequence is read in triplets (codons) until a stop codon is encountered. If a stop codon is not present within the sequence, the ORF is considered to extend to the end of the sequence.
Analysis:
First Start Codon: ATG at position 1:
- Sequence from ATG:
ATGCATGCTAGCTAATGCCCGGGTAACCCATGA - Codon Breakdown:
- Stop Codons Encountered:
- TAA at position 13
- Identified ORFs:
- ORF1: ATG CAT GCT AGC TAA
- Start: ATG (positions 1-3)
- Stop: TAA (positions 13-15)
- Length: 15 nucleotides (5 codons)
- ORF1: ATG CAT GCT AGC TAA
Second Start Codon: ATG at position 5:
- Sequence from ATG:
ATGCTAGCTAATGCCCGGGTAACCCATGA - Codon Breakdown:
- Stop Codon Encountered:
- No stop codon
Third Start Codon: ATG at position 14:
- Sequence from ATG:
ATGCCCGGGTAACCCATGA - Codon Breakdown:
- Stop Codon Encountered:
- TAA at position at 23
- Identified ORFs:
- ORF2: ATG CCC GGG TAA
- Start: ATG (positions 21-23)
- Stop: TAA (positions 27-29)
- Length: 15 nucleotides (5 codons)
- ORF2: ATG CCC GGG TAA
Summary of ORFs on Forward Strand:
- ORF1: ATG CAT GCT AGC TAA (15 nt)
- ORF2: ATG CCC GGG TAA (12 nt)
Identifying ORFs on the Reverse Complement Strand
To analyze the reverse complement strand, we first generate its complementary sequence and reverse it to maintain the 5' to 3' orientation.
Reverse Complement Strand:
Original: 5' A T G C A T G C T A G C T A A T G C C C G G G T A A C C C A T G A 3'
Complement: 3' T A C G T A C G A T C G A T T A C G G G C C C A T T G G G T A C T 5'
- Reversing to 5' to 3':
Analysis:
First Start Codon: ATG at position 3
- Sequence from ATG:
ATGGGTTACCCGGGCATTAGCTAGCATGCAT - Codon Breakdown:
- Stop Codon Encountered:
- TAG at position at 24
Second Start Codon: ATG at position 24
- Sequence from ATG:
ATGCAT - Codon Breakdown:
- Stop Codon Encountered:
- No complete stop codon within the sequence.
Summary of ORFs on Reverse Complement Strand:
- ORF3: ATG GGT TAC CCG GGC ATT AGC TAG (24 nt)
Consolidated List of Identified ORFs
Combining the findings from both strands, the complete list of ORFs in the given bacterial DNA sequence is as follows:
- ORF1 (Forward Strand):
ATG CAT GCT AGC TAA - ORF2 (Forward Strand):
ATG CCC GGG TAA - ORF3 (Reverse Strand):
ATG GGT TAC CCG GGC ATT AGC TAG
Determining the Most Likely Real Gene
Among the identified ORFs, determining which one is most likely to represent a genuine gene involves evaluating several criteria. The primary factors to consider include:
- ORF Length:
- Longer ORFs: Generally more likely to encode functional proteins, as they have sufficient length to translate into meaningful polypeptides.
- Shorter ORFs: May represent non-coding regions, regulatory elements, or be artifacts of the sequence analysis.
- Presence of Complete Codon Structure:
- Start and Stop Codons: ORFs with both start and stop codons are more likely to represent actual genes.
- Incomplete ORFs: Lack of a stop codon may indicate partial gene sequences or pseudogenes.
- Codon Usage and Conservation:
- Consistent Codon Usage: Genes typically exhibit codon usage patterns that match the organism’s preferred codon usage.
- Conservation Across Species: Genes conserved across related species are more likely to be functional.
- Functional Motifs and Domains:
- Protein Domains: Presence of known protein domains or motifs within the ORF supports its candidacy as a gene.
- Regulatory Elements: Association with regulatory sequences such as promoters enhances the likelihood of gene function.
- Contextual Genomic Features:
- Ribosomal Binding Sites (RBS): Proximal RBS motifs upstream of the start codon are indicative of functional genes.
- Operon Structure: Genes organized within operons often function together, providing additional context for gene identification.
Applying the Criteria to Identified ORFs:
- ORF1:
- Length: 5 codons
- Features: Contains both start (ATG) and stop (TAA) codons.
- Assessment: Relatively short; may represent a partial gene or a small peptide-coding sequence.
- ORF2:
- Length: 4 codons
- Features: Contains both start (ATG) and stop (TAA) codons.
- Assessment: Shorter than ORF1.
- ORF3:
- Length: 8 codons
- Features: Contains both start (ATG) and stop (TAG) codons.
- Assessment: Longer than ORF1 in length and features; may represent a small gene or regulatory element.
Conclusion:
Among the identified ORFs, ORF3 emerges as the most promising candidate for a real gene due to its longer length. While bacterial genes are typically longer to encode functional proteins, the short length of ORF3 (24 nucleotides) suggests it may encode a small peptide or represent a partial gene sequence. To further validate ORF3 as a genuine gene, additional analyses could be performed, such as:
- Codon Usage Analysis: Comparing the codon usage of ORF3 to known bacterial genes to assess consistency.
- Functional Annotation: Searching for homologous sequences in protein databases to identify potential functions.
- Presence of RBS: Verifying the presence of a ribosomal binding site upstream of ORF3 to support its role in translation initiation.
- Experimental Validation: Conducting laboratory experiments, such as gene expression assays, to confirm the functionality of the ORF.
Overall, while ORF3 stands out as the most likely real gene based on the provided sequence and initial analysis, comprehensive validation incorporating multiple lines of evidence is essential to confirm its authenticity.
b) Compare and contrast the challenges of identifying genes in GC-rich vs. AT-rich prokaryotic genomes. How might these differences affect gene prediction strategies?
Solution
Gene identification in prokaryotic genomes is a critical step in understanding the functional capabilities of microorganisms. Prokaryotic genomes exhibit a wide range of guanine-cytosine (GC) content, influencing various genomic features and posing distinct challenges in gene prediction. GC-rich and AT-rich genomes represent the two extremes of this spectrum, each introducing unique obstacles for accurate gene identification. This discussion compares the challenges associated with identifying genes in GC-rich versus AT-rich prokaryotic genomes and examines how these differences influence gene prediction strategies.
Challenges in GC-Rich Prokaryotic Genomes
- Codon Usage Bias:
- Impact on Gene Prediction: High GC content leads to a preference for codons rich in guanine (G) and cytosine (C). This skewed codon usage can confound gene prediction algorithms that rely on standard codon frequencies derived from model organisms with average GC content.
- Example: In GC-rich genomes, codons like GCG (Alanine) are used more frequently than their synonymous counterparts with lower GC content, altering the expected patterns of coding sequences.
- Atypical Start and Stop Codons:
- Impact on Gene Boundary Identification: The prevalence of GC-rich codons can reduce the frequency of typical start (AUG) and stop codons (UAA, UAG, UGA), making it difficult to accurately define gene boundaries.
- Alternative Codons: GC-rich genomes may utilize non-standard start codons (e.g., GUG or UUG) more frequently, necessitating adjustments in prediction algorithms to recognize these signals.
- Secondary Structures Affecting Sequencing:
- Impact on Sequence Quality: High GC content can lead to stable secondary structures like hairpins in the DNA, which impede DNA polymerase during sequencing and PCR amplification, resulting in poor-quality reads or sequencing gaps.
- Consequence for Gene Identification: These sequencing challenges can lead to incomplete assemblies, making it difficult to identify genes within these problematic regions.
- Compressed Genomes:
- Short Intergenic Regions: GC-rich genomes often have compact genomes with minimal non-coding sequences, reducing the distinctiveness between coding and non-coding regions.
- Overlap of Genes: The proximity of genes can result in overlapping coding sequences, complicating the prediction of individual genes.
Challenges in AT-Rich Prokaryotic Genomes
- Homopolymeric Runs and Repeats:
- Impact on Sequencing and Assembly: AT-rich genomes frequently contain long stretches of adenine (A) or thymine (T) bases and simple sequence repeats, which can cause slippage during sequencing and errors in read alignment.
- Assembly Errors: These repetitive regions can lead to misassemblies or gaps, hindering accurate gene identification.
- Codon Usage Bias:
- Impact on Gene Prediction: AT-rich genomes prefer codons with more A and T nucleotides, which can differ significantly from the codon usage in standard models, leading to reduced sensitivity and specificity in gene prediction.
- Example: Codons like AAA (Lysine) are used more frequently, altering the expected nucleotide patterns in coding regions.
- Reduced Signal Recognition:
- Promoter and Regulatory Elements: AT-rich genomes may have less conserved promoter regions and ribosome binding sites, which are typically GC-rich. This can make it harder for algorithms to detect these regulatory signals.
- Impact on Gene Start Sites: The ambiguity in promoter regions affects the accurate prediction of transcription start sites.
- Abundant Non-coding DNA:
- Long Intergenic Regions: AT-rich genomes may have larger intergenic spaces, increasing the difficulty of distinguishing between coding and non-coding regions due to the presence of non-functional open reading frames (ORFs).
- Pseudogenes and Mobile Elements: Higher prevalence of pseudogenes and transposable elements can lead to false positives in gene prediction.
Comparative Analysis of Challenges
- Codon Bias Differences:
- GC-Rich Genomes: Exhibit a bias towards codons with G and C, affecting the expected patterns in coding sequences.
- AT-Rich Genomes: Prefer codons with A and T, requiring adjustments in prediction models to account for this bias.
- Sequencing Difficulties:
- GC-Rich Genomes: Secondary structures can hinder sequencing, leading to incomplete data.
- AT-Rich Genomes: Homopolymer runs can cause sequencing errors and assembly problems.
- Regulatory Signal Detection:
- GC-Rich Genomes: May have atypical start/stop codons and compressed intergenic regions.
- AT-Rich Genomes: Less conserved promoter regions and regulatory elements reduce signal recognition accuracy.
- Genome Structure:
- GC-Rich Genomes: Tend to have more compact genomes with overlapping genes.
- AT-Rich Genomes: Often contain larger non-coding regions and repetitive elements.
Impact on Gene Prediction Strategies
- Algorithm Customization:
- Codon Usage Models: Gene prediction tools must incorporate organism-specific codon usage tables reflecting the nucleotide composition bias to improve accuracy.
- Training Data Sets: Algorithms should be trained on genomes with similar GC content to capture the unique features of gene structure.
- Signal Recognition Adjustments:
- Alternative Start Codons: Include recognition of non-standard start codons prevalent in GC-rich genomes.
- Promoter Prediction Models: Adapt models to detect less conserved or AT-rich promoter regions in AT-rich genomes.
- Handling Sequencing Errors:
- Improved Sequencing Techniques: Employ sequencing technologies that reduce errors in homopolymer regions for AT-rich genomes (e.g., single-molecule real-time sequencing).
- Error Correction Algorithms: Use computational methods to correct sequencing errors associated with GC-rich secondary structures.
- Genome Assembly Strategies:
- Assembly Algorithms: Utilize assemblers optimized for high GC or AT content to mitigate assembly errors due to nucleotide composition biases.
- Read Coverage Analysis: Implement strategies to identify and resolve regions with uneven coverage caused by sequencing biases.
- Annotation Pipelines:
- Dynamic Parameter Adjustment: Gene prediction pipelines should dynamically adjust parameters based on the GC content of the genome under study.
- Integration of Multiple Tools: Combine results from different gene prediction tools to compensate for the limitations of each in handling extreme GC or AT content.
- Use of Comparative Genomics:
- Ortholog Identification: Leverage evolutionary conservation by comparing with closely related organisms to identify genes that might be missed due to nucleotide bias.
- Phylogenetic Profiles: Use profiles to predict gene function and presence, aiding in the identification of genes obscured by GC or AT content.
Q09¶
In gene prediction, coding scores are often used to identify potential protein-coding regions. One method to calculate coding scores involves analyzing the frequency of hexamers (6-nucleotide sequences) in known genes versus the entire genome. Given the following information:
- Hexamer:
ATGGCC - Frequency in known genes (\(G\)): 0.015
- Frequency in the whole genome (\(B\)): 0.005
a) Calculate the coding score \(C(w)\) for this hexamer using the formula:
Where \(G(w)\) is the frequency of the word in genes, and \(B(w)\) is the frequency in the whole genome.
Warning
The prodigal paper was unclear what base the \(\log\) was in, but the C code uses the built-in log function which is the natural logarithm.
I will accept \(\log_{10}\) as well just for this problem.
Solution
The coding score \( C(w) \) quantifies how much more (or less) frequently a particular hexamer occurs in known genes compared to the entire genome. It is calculated using the formula:
Where:
- \( G(w) \) is the frequency of the hexamer in known genes.
- \( B(w) \) is the frequency of the hexamer in the whole genome.
- \( \ln \) denotes the natural logarithm (base \( e \)).
Given:
- \( G(w) = 0.015 \)
- \( B(w) = 0.005 \)
First, compute the ratio \( \frac{G(w)}{B(w)} \):
Next, calculate \( C(w) \):
Answer:
The coding score \( C(w) \) for the hexamer ATGGCC is approximately 1.0986.
(If you used \(\log_{10}\), this would approximatley be 0.477.)
b) Interpret the result. What does the score indicate about this hexamer?
Solution
The coding score indicates how much more likely a hexamer is to be found in coding regions compared to the genome overall:
- Positive Score (\( C(w) > 0 \)): The hexamer is more frequent in coding regions.
- Negative Score (\( C(w) < 0 \)): The hexamer is less frequent in coding regions.
- Zero Score (\( C(w) = 0 \)): The hexamer is equally frequent in both regions.
For ATGGCC:
- \( C(w) \approx 1.0986 \): A positive value, indicating higher frequency in coding regions.
- The ratio \( \frac{G(w)}{B(w)} = 3 \) means the hexamer is three times more common in known genes than in the genome overall.
The positive coding score suggests that ATGGCC is significantly associated with protein-coding regions, making it a potential indicator of coding sequences.
c) If you found this hexamer in an uncharacterized region of DNA, how might this score influence your assessment of whether this region is likely to be protein-coding?
Solution
If the hexamer ATGGCC is found in an uncharacterized DNA region, the positive coding score has the following implications:
- Increased Likelihood of Coding Potential: The region is more likely to be protein-coding because it contains a hexamer that is prevalent in known genes.
- Support for Gene Prediction: The presence of such hexamers can be used as evidence in computational gene prediction algorithms to identify potential genes.
The positive coding score of ATGGCC would lead you to favor the hypothesis that the uncharacterized DNA region is part of a protein-coding gene. This score strengthens the assessment by providing statistical evidence that the region shares characteristics with known coding sequences.
Q10¶
You are analyzing a short DNA sequence to determine if there is likely to be a protein-coding gene.
- Given DNA sequence:
ATATGCATGCTTAGCTTA
You have a table of pre-calculated coding scores for all possible hexamers based on known genes in the bacteria's genome.
| Hexamer | Score | Hexamer | Score |
|---|---|---|---|
| ATATGC | -0.2 | CATGCT | 1.2 |
| TATGCA | -0.4 | ATGCTT | 0.7 |
| ATGCAT | 0.8 | TGCTTA | -0.1 |
| TGCATG | -0.2 | GCTTAG | 0.3 |
| GCATGC | 0.5 | CTTAGC | 0.6 |
| TTAGCT | -0.3 | TAGCTT | -0.5 |
| AGCTTA | 0.1 |
a) Calculate the overall coding score for the gene in this DNA sequence. To do this:
- Determine the score for each overlapping hexamer in the gene.
- Sum these scores to get the total coding score for the gene.
Remember that you shift over one codon a time. (I had to read their C code to figure this out.)
Solution
In gene prediction, one of the key steps is to determine whether a DNA sequence is likely to be a protein-coding gene. This involves:
- Identifying an open reading frame (ORF) with a start codon and a stop codon in the same reading frame.
- Calculating the overall coding score by analyzing hexamer frequencies within the ORF.
- Interpreting the coding score to assess the likelihood of the sequence being protein-coding.
In this walkthrough, we will apply these steps to the given DNA sequence using the provided table of hexamer coding scores.
Given DNA Sequence
Identify the Open Reading Frame (ORF)
An open reading frame (ORF) is a sequence of DNA that could potentially encode a protein. It starts with a start codon and ends with a stop codon, with all codons read in the same frame.
- Start Codon: ATG
- Stop Codons: TAA, TAG, TGA
Analyzing the Three Possible Reading Frames
We will examine all three possible reading frames to find an ORF.
Reading Frame 1:
Positions: 1-3, 4-6, 7-9, 10-12, 13-15, 16-18
Codon 1 (1-3): A T A
Codon 2 (4-6): T G C
Codon 3 (7-9): A T G (Start Codon)
Codon 4 (10-12): C T T
Codon 5 (13-15): A G C
Codon 6 (16-18): T T A
- No Stop Codon found in this frame.
- Start Codon at Codon 3 (positions 7-9).
Reading Frame 2:
Positions: 2-4, 5-7, 8-10, 11-13, 14-16, 17-18
Codon 1 (2-4): T A T
Codon 2 (5-7): G C A
Codon 3 (8-10): T G C
Codon 4 (11-13): T T A
Codon 5 (14-16): G C T
Codon 6 (17-18): T A (Incomplete)
- No Start or Stop Codon found in this frame.
Reading Frame 3:
Positions: 3-5, 6-8, 9-11, 12-14, 15-17
Codon 1 (3-5): A T G (Start Codon)
Codon 2 (6-8): C A T
Codon 3 (9-11): G C T
Codon 4 (12-14): T A G (Stop Codon)
Codon 5 (15-17): C T T
- Start Codon at Codon 1 (positions 3-5).
- Stop Codon at Codon 4 (positions 12-14).
- ORF Length: From Codon 1 to Codon 4.
Conclusion: We have identified an ORF in Reading Frame 3 starting with a start codon (ATG) and ending with a stop codon (TAG) in the same frame.
Extract Hexamers Within the ORF
Now that we have the ORF, we need to extract hexamers (6-nucleotide sequences) within it.
- Shift over one codon (three nucleotides) at a time.
- This ensures that the hexamers align with the reading frame and reflect codon usage.
ORF Nucleotide Sequence (Positions 3-14):
- Hexamer 1 (Positions 3-8):
ATGCAT - Hexamer 2 (Positions 6-11):
CATGCT - Hexamer 3 (Positions 9-14):
GCTTAG
Retrieve Coding Scores for Each Hexamer
Using the provided coding scores table:
| Hexamer | Score |
|---|---|
| ATGCAT | 0.8 |
| CATGCT | 1.2 |
| GCTTAG | 0.3 |
Calculate the Total Coding Score
The total coding score is the sum of the scores of all hexamers within the ORF.
b) What is the average coding score per hexamer for this sequence?
Solution
Identify the Number of Hexamers
From the analysis of the open reading frame (ORF), we have the following hexamers:
- Hexamer 1: ATGCAT (Score: 0.8)
- Hexamer 2: CATGCT (Score: 1.2)
- Hexamer 3: GCTTAG (Score: 0.3)
Total Number of Hexamers: 3
Recall the Total Coding Score
The sum of the coding scores for these hexamers is:
Calculate the Average Coding Score per Hexamer
The average coding score per hexamer is calculated by dividing the total coding score by the number of hexamers:
Plugging in the numbers:
Answer:
The average coding score per hexamer for this sequence is approximately 0.7667.
- Positive Average Score: An average coding score per hexamer greater than zero indicates that, on average, the hexamers within the sequence are characteristic of protein-coding regions.
- Strength of Coding Potential: An average score of approximately 0.7667 suggests a strong coding potential for the sequence.
- Consistency Across Hexamers: The relatively high average reflects that most hexamers contribute positively to the overall coding score.
c) Based on your results, do you think this gene is likely to be protein-coding? Explain your reasoning.
Solution
Yes, based on the results of the analysis, it is highly likely that this gene is protein-coding
1. Identification of an Open Reading Frame (ORF):
- Start Codon: The sequence contains a start codon (ATG) at positions 3-5 in Reading Frame 3.
- Stop Codon: A stop codon (TAG) is present at positions 12-14 in the same reading frame.
- Continuous Reading Frame: The codons between the start and stop codons are in-frame, allowing for uninterrupted translation.
- Significance: The presence of an ORF with both start and stop codons is a key indicator of a protein-coding gene.
2. Positive Total Coding Score:
- Calculated Total Coding Score: The sum of the coding scores for the hexamers within the ORF is 2.3.
- Interpretation: A positive total coding score suggests that the sequence has characteristics similar to known protein-coding regions.
- Strength of Evidence: A score of 2.3 is significantly positive, indicating strong coding potential.
3. High Average Coding Score per Hexamer:
- Average Score: The average coding score per hexamer is approximately 0.7667.
- Consistent Positive Scores: All hexamers within the ORF have positive scores, reinforcing the coding potential.
- Implication: A high average score per hexamer suggests that, on average, the sequence closely resembles coding sequences in terms of hexamer composition.
4. Individual Hexamer Scores Support Coding Potential:
- Hexamer 1 (ATGCAT): Score 0.8
- Hexamer 2 (CATGCT): Score 1.2
- Hexamer 3 (GCTTAG): Score 0.3
- Analysis: Each hexamer has a positive coding score, indicating that these sequences are more frequently found in known genes compared to non-coding regions.
- Cumulative Effect: The positive contributions of each hexamer strengthen the overall evidence for coding potential.
5. Alignment with Known Gene Features:
- Reading Frame Integrity: The codons are properly aligned without frameshifts, which is essential for correct protein translation.
- Codon Usage: The sequence likely follows typical codon usage patterns observed in the organism's protein-coding genes.
- Biological Plausibility: The structure of the sequence is consistent with what is expected for a functional gene.
6. Absence of Negative Indicators:
- No Premature Stop Codons: There are no in-frame stop codons before the identified stop codon, which could truncate the protein.
- No Significant Negative Coding Scores: Negative scores can indicate non-coding regions; their absence supports the coding potential.
- Sequence Length: The length of the ORF is sufficient to encode a peptide of reasonable size, which is typical for bacterial genes.
7. Statistical Evidence from Coding Scores:
- Methodology Validity: Coding scores are based on statistical analyses comparing hexamer frequencies in known genes versus the whole genome.
- Reliability: The positive scores provide quantitative support for the sequence being protein-coding.
- Comparative Analysis: The sequence aligns well with known coding patterns in the bacterial genome.
d) Discuss potential limitations of using this hexamer-based coding score approach for gene prediction. What factors might lead to false positives or false negatives?
Solution
Hexamer-based coding score methods are widely utilized in gene prediction algorithms due to their ability to capture local sequence patterns characteristic of protein-coding regions. By analyzing the frequency of six-nucleotide sequences (hexamers) in known genes versus the entire genome, these methods provide statistical measures to identify potential coding regions. However, despite their utility, hexamer-based approaches possess several limitations that can lead to inaccuracies in gene prediction. This comprehensive discussion outlines the primary limitations and factors that may result in false positives and false negatives.
Dependence on Existing Gene Models
Hexamer-based methods rely heavily on the frequency of hexamers observed in known genes. This dependence means that the effectiveness of the approach is contingent on the quality and comprehensiveness of the training data.
- False Negatives: Novel genes or genes with atypical hexamer compositions not represented in the training set may be overlooked.
- False Positives: Regions with hexamer patterns similar to known genes but not actually coding may be incorrectly identified as genes.
A newly discovered gene with unique regulatory elements or unusual codon usage may not align well with existing hexamer profiles, causing it to be missed by prediction algorithms.
Codon Bias and Genome Composition Variability
Different organisms exhibit varying codon biases and nucleotide compositions. Hexamer frequencies are influenced by these biases, and extreme GC-rich or AT-rich genomes may present challenges.
- False Positives in GC/AT-Rich Genomes: In genomes with extreme nucleotide compositions, certain hexamers may appear more frequently in non-coding regions purely due to base composition, leading to erroneous gene predictions.
- False Negatives: Genuine coding regions in such genomes may have hexamer patterns that diverge from typical coding regions in more balanced genomes, causing them to be missed.
A GC-rich genome may have stable secondary structures or specific codon usage that affects hexamer frequencies, complicating the distinction between coding and non-coding regions.
Overlapping Genes and Operonic Structures
Prokaryotic genomes often contain overlapping genes and operonic structures, where multiple genes share regions of DNA.
- False Positives: Overlapping genes may create complex hexamer patterns that resemble coding regions, even if only part of the sequence is genuinely coding.
- False Negatives: Genes overlapping with non-coding regions or other genes may dilute the hexamer signal, making it harder to detect.
An operon with tightly packed genes may exhibit combined hexamer frequencies that do not accurately reflect individual gene coding potentials.
Short Genes and Insufficient Hexamer Data
Short genes or small open reading frames (sORFs) provide limited hexamer data, reducing the statistical power of the coding score.
- False Negatives: Short coding sequences may not accumulate enough positive hexamer scores to surpass prediction thresholds.
- False Positives: Limited hexamer data can make it easier for non-coding regions to coincidentally exhibit high coding scores.
A small peptide-coding gene with only a few hexamers may not receive a sufficiently high total coding score to be confidently identified as coding.
Pseudogenes and Mobile Genetic Elements
Pseudogenes and mobile genetic elements (e.g., transposons, plasmids) can contain hexamer patterns similar to functional genes.
- False Positives: These non-functional or mobile elements may be incorrectly predicted as protein-coding genes due to their similar hexamer compositions.
- False Negatives: Conversely, fragmented pseudogenes may disrupt hexamer patterns, making true coding regions harder to detect.
A transposable element with conserved coding sequences for transposase enzymes may be mistakenly annotated as a separate gene.
Regulatory Regions and Non-Coding RNAs
Some regulatory regions and non-coding RNAs (ncRNAs) possess hexamer compositions that overlap with those of protein-coding genes.
- False Positives: Regulatory elements like promoters, enhancers, or ncRNAs may exhibit hexamer frequencies akin to coding regions, leading to incorrect gene predictions.
- False Negatives: Genuine coding regions adjacent to regulatory elements might have diluted hexamer signals.
An enhancer region with high GC content may generate hexamer patterns that resemble coding regions, causing misannotation.
Sequencing Errors and Genome Assembly Issues
Errors introduced during DNA sequencing or genome assembly can distort the true hexamer frequencies.
- False Positives: Sequencing errors may create artificial hexamers with high coding scores.
- False Negatives: Errors that disrupt genuine hexamer patterns can prevent accurate gene prediction.
Insertion or deletion errors leading to frameshifts can alter hexamer sequences, affecting the overall coding score calculation.
Context-Dependence and Higher-Order Sequence Features
Hexamer-based methods focus on local sequence patterns and may overlook broader contextual or higher-order sequence features essential for accurate gene prediction.
- False Positives: Lack of consideration for gene context (e.g., genomic location, regulatory context) may result in mispredictions.
- False Negatives: Ignoring larger sequence motifs or structural features can lead to missed coding regions.
A hexamer pattern typical of coding regions but located within a known non-coding genomic island may be incorrectly predicted as a gene.
Alternative Start Codons and Non-Standard Coding Patterns
Some genes utilize non-standard start codons or have atypical coding patterns that are not well-represented in the hexamer training data.
- False Negatives: Genes with alternative start codons (e.g., GTG, TTG) may not trigger high coding scores if such codons are not accounted for.
- False Positives: Misinterpretation of alternative codon usages in non-coding regions may lead to incorrect gene predictions.
A gene starting with GTG, a less common start codon, may not be recognized as coding if the hexamer frequencies do not account for such variations.
Insensitivity to Longer Sequence Patterns and Gene Structure
Hexamer-based approaches analyze fixed-length sequences and may miss longer or more complex sequence patterns essential for gene structure recognition, such as splice sites in eukaryotes (though less relevant for prokaryotes).
- False Negatives: Complex gene structures may not be captured, leading to missed predictions.
- False Positives: Overreliance on short sequence motifs without structural context can cause mispredictions.
In prokaryotes, operon structures and regulatory sequences extending beyond hexamer boundaries may influence gene expression but remain undetected by hexamer-based scores.
Evolutionary Conservation and Functional Divergence
Hexamer-based methods may not account for evolutionary conservation or functional divergence among genes, leading to mismatches in coding score expectations.
- False Negatives: Highly conserved genes with unique hexamer compositions may be underrepresented in training data, reducing prediction accuracy.
- False Positives: Conserved non-coding regions may exhibit similar hexamer patterns to coding regions, causing incorrect predictions.
Essential genes with highly conserved sequences across species may present hexamer patterns that diverge from the training set, affecting prediction algorithms.
Computational and Statistical Limitations
Hexamer-based approaches involve statistical modeling that may suffer from limitations such as insufficient sample sizes, overfitting, or assumptions about hexamer independence.
- False Positives: Overfitting to training data can cause the model to predict genes where none exist.
- False Negatives: Simplistic statistical models may fail to generalize to diverse genomic contexts.
Assuming independence between hexamers ignores the potential correlations and dependencies in genomic sequences, reducing the model's predictive power.
Strategies to Mitigate Limitations
While hexamer-based coding score methods have inherent limitations, several strategies can enhance their accuracy:
- Integrate Multiple Features: Combine hexamer scores with other genomic features such as promoter motifs, ribosome binding sites, and gene conservation.
- Use Machine Learning Models: Employ advanced machine learning techniques that can capture higher-order dependencies and contextual information.
- Adapt to Genome Composition: Customize hexamer training sets to reflect the specific nucleotide composition and codon bias of the target organism.
- Incorporate Comparative Genomics: Leverage evolutionary conservation by comparing sequences across related species to validate gene predictions.
- Enhance Training Data: Expand and diversify training datasets to include a wide range of gene types, including those with non-standard features.
Programming+¶
These problems are not required and will not impact your BIOSC 1540 grade. The instructor will assess these separately to validate correctness without an assigned grade. Thus, you may work on these problems individually or in a team-based setting and "due" by the end of the semester. Happy coding!
Acceptable languages: Python v3.10+, Rust v1.80+, Mojo v24.4+
Rewards
Engaging with these optional programming problems offers several valuable academic and professional growth opportunities.
- Consistent engagement with these Programming+ problems will allow me to write more detailed, compelling recommendation letters highlighting your computational skills. These personalized letters can significantly boost your applications for future academic programs, internships, or job opportunities.
- If there is enough interest, optional Friday recitations will be provided. This will give you individualized attention to accelerate learning and actionable feedback on your code and problem-solving approaches.
- Exceptional solutions may be featured on our course website with the students' permission. This is a way for us to recognize and appreciate your hard work and dedication to these problems.
Note
These problems would be similar to ones given in a major-only version of the class. Although, there would be more relevant instructions during class and would be given more than a week to complete.
P01
Implement the Needleman-Wunsch algorithm for global sequence alignment.
a) Write a function needleman_wunsch(seq1, seq2, match_score, mismatch_score, gap_penalty) that takes two sequences and scoring parameters as input and returns the optimal alignment score and the aligned sequences.
b) Test your implementation with the sequences from Q04 in the assignment:
ATGCACTAGCTAATGCTACGTA
Use a match score of +1, mismatch score of -1, and gap penalty of -2.
c) Add functionality to visualize the scoring matrix and the traceback path.
P02
Implement the Smith-Waterman algorithm for local sequence alignment.
a) Write a function smith_waterman(seq1, seq2, match_score, mismatch_score, gap_penalty) that takes two sequences and scoring parameters as input and returns the optimal local alignment score and the aligned subsequences.
b) Test your implementation with the same sequences from P01.
c) Add functionality to visualize the scoring matrix and the traceback path, highlighting the highest-scoring local alignment.
P03
Create a script to find all possible Open Reading Frames (ORFs) in a given DNA sequence.
a) Write a function find_orfs(sequence, min_length=100) that takes a DNA sequence as input and returns a list of all ORFs longer than the minimum length. Consider both strands of the DNA.
b) Implement a function to translate the ORFs into amino acid sequences.
c) Test your implementation with the sequence from Q08 in the assignment: 5' ATGCATGCTAGCTAATGCCCGGGTAACCCATGA 3'
d) Add functionality to identify the longest ORF and potential start codons (ATG) within the ORFs.
P04
Implement a Python script to calculate coding scores for a DNA sequence based on hexamer frequencies.
a) Write a function calculate_coding_score(sequence) that takes a DNA sequence and returns the overall coding score for the sequence.
Use the following function to compute the hexamer scores.
def compute_hexamer_score(hexamer):
"""
Compute a score for a given hexamer based on its nucleotide composition.
This function uses a simple scoring system based on GC content and specific dinucleotide frequencies.
Args:
hexamer (str): A string of 6 nucleotides (A, T, C, or G)
Returns:
The computed score for the hexamer
"""
if len(hexamer) != 6 or not all(base in 'ATCG' for base in hexamer.upper()):
raise ValueError("Input must be a string of 6 nucleotides (A, T, C, or G)")
hexamer = hexamer.upper()
# Base scores
base_scores = {'A': 1, 'T': 1, 'C': 2, 'G': 2}
# Dinucleotide bonus scores
dinucleotide_bonus = {'CG': 0.5, 'GC': 0.5, 'AT': -0.3, 'TA': -0.3}
# Calculate base score
score = sum(base_scores[base] for base in hexamer)
# Add dinucleotide bonuses
for i in range(5):
dinucleotide = hexamer[i:i+2]
if dinucleotide in dinucleotide_bonus:
score += dinucleotide_bonus[dinucleotide]
# Normalize score
score = score / 6
return round(score, 2)
# Example usage:
print(compute_hexamer_score('ATGCAT')) # Output: 1.62
print(compute_hexamer_score('CGCGCG')) # Output: 2.5
print(compute_hexamer_score('ATATAG')) # Output: 0.95
b) Implement the sliding window approach, shifting by one codon (3 nucleotides) at a time, as mentioned in Q10 of the assignment.
c) Test your implementation with the sequence and hexamer scores from Q10.
A04
Q01¶
Compare and contrast genomics and transcriptomics in terms of:
- The type of information they provide;
- The frequency of sampling required;
- Their ability to capture cellular responses.
Solution
In the field of molecular biology, genomics and transcriptomics are two pivotal disciplines that offer distinct yet complementary insights into the functioning of living organisms. While both are integral to understanding biological systems at the molecular level, they differ fundamentally in the type of information they provide, the frequency of sampling they necessitate, and their capacity to capture cellular responses.
| Aspect | Genomics | Transcriptomics |
|---|---|---|
| Information Provided | Complete DNA sequence, gene locations, structural variations | Gene expression levels, splice variants, non-coding RNA expression |
| Stability | Largely static across an organism's lifespan | Highly dynamic, varies with time, conditions, and cell states |
| Sampling Frequency | Infrequent; typically one-time per individual or species | Frequent and time-resolved; multiple samples under different conditions |
| Cellular Response Capture | Limited; provides potential response capabilities based on genetic makeup | Comprehensive; captures real-time and dynamic changes in gene expression |
| Applications | Identifying genetic predispositions, evolutionary studies, personalized medicine | Understanding gene regulation, cellular responses, disease mechanisms, developmental processes |
| Technological Methods | Whole-genome sequencing, SNP genotyping, comparative genomics | RNA sequencing (RNA-Seq), microarrays, single-cell transcriptomics |
Q02¶
Compare RNA-seq to microarray technology. What are the key advantages of RNA-seq?
Solution
While microarray technology laid the groundwork for large-scale gene expression studies, RNA-Seq has emerged as a more powerful and versatile tool, offering comprehensive, sensitive, and precise insights into the transcriptome. Its ability to detect novel transcripts, provide higher dynamic range and specificity, and facilitate a deeper understanding of gene regulation positions RNA-Seq as the technology of choice for modern genomics research. As sequencing technologies continue to advance, the advantages of RNA-Seq are likely to further solidify its role in unraveling the complexities of gene expression and regulation.
Overview of Microarray Technology and RNA-Seq
Microarray Technology: Microarrays, introduced in the mid-1990s, revolutionized gene expression studies by allowing simultaneous measurement of thousands of genes. A microarray consists of a solid surface, typically a glass slide, onto which probes (short DNA sequences) corresponding to specific genes are fixed in an orderly grid. During an experiment, fluorescently labeled cDNA or cRNA derived from the sample's RNA is hybridized to the probes on the array. The intensity of fluorescence at each probe spot correlates with the expression level of the corresponding gene.
RNA Sequencing (RNA-Seq): RNA-Seq, emerging in the late 2000s, leverages next-generation sequencing (NGS) technologies to provide a comprehensive and high-resolution view of the transcriptome. Unlike microarrays, RNA-Seq does not rely on predefined probes. Instead, it sequences the entire pool of RNA molecules (after conversion to cDNA) present in a sample, generating millions of short reads that are then aligned to a reference genome or assembled de novo. This approach enables both the quantification of gene expression and the discovery of novel transcripts, alternative splicing events, and other transcriptomic complexities.
Comparative Analysis: RNA-Seq vs. Microarrays
| Feature | Microarrays | RNA-Seq |
|---|---|---|
| Detection Method | Hybridization of labeled RNA to fixed probes | Sequencing of cDNA fragments |
| Dynamic Range | Limited (typically 2-3 orders of magnitude) | Broad (up to 6-7 orders of magnitude) |
| Sensitivity | Moderate; may miss low-abundance transcripts | High; can detect lowly expressed genes and rare transcripts |
| Specificity | Dependent on probe design; potential cross-hybridization issues | High; sequence-based, reducing cross-reactivity |
| Coverage | Limited to known genes and transcripts defined by probe set | Comprehensive; includes known and novel transcripts |
| Resolution | Gene-level; limited ability to detect isoforms | Nucleotide-level; can identify alternative splicing and variants |
| Quantification | Relative expression levels based on fluorescence intensity | Absolute expression levels based on read counts |
| Cost | Generally lower per sample for large-scale studies | Initially higher, but costs have been decreasing |
| Data Analysis Complexity | Relatively simpler bioinformatics requirements | More complex, requiring advanced bioinformatics tools |
| Reproducibility | High within the same platform | High, with consistency across different sequencing runs |
| Scalability | Limited by the need for specific probes | Highly scalable with advancements in sequencing technologies |
Key Advantages of RNA-Seq Over Microarrays
While both technologies have their merits, RNA-Seq offers several distinct advantages that make it the preferred choice for many contemporary gene expression studies:
Comprehensive Transcriptome Coverage
- RNA-Seq does not rely on predefined probes, allowing the detection of previously unknown genes, non-coding RNAs, and novel splice variants. This capability is crucial for expanding our understanding of the transcriptomic landscape beyond what is currently annotated.
- RNA-Seq can identify and quantify different splice variants of genes, providing insights into the complexity of gene regulation and the functional diversity of proteins. Microarrays, limited by their probe design, often cannot distinguish between isoforms.
Enhanced Sensitivity and Dynamic Range
- RNA-Seq's high sensitivity enables the detection of transcripts expressed at very low levels, which microarrays may fail to identify due to background noise and limited dynamic range.
- RNA-Seq can accurately quantify gene expression over a broad range of abundances, from very low to extremely high, without saturation issues inherent in microarray fluorescence measurements.
Higher Specificity and Accuracy
- RNA-Seq relies on direct sequencing of RNA-derived cDNA, minimizing cross-hybridization issues that can occur with microarrays. This leads to more accurate and specific identification of transcripts.
- RNA-Seq provides nucleotide-level resolution, allowing for the detection of single nucleotide polymorphisms (SNPs), insertions, deletions, and other genetic variations within transcripts. Microarrays generally lack this level of detail.
Flexibility and Adaptability
- RNA-Seq does not require the design and optimization of probes for each gene, making it more adaptable to studies involving non-model organisms or organisms with poorly annotated genomes.
- RNA-Seq can identify gene fusions, structural rearrangements, and other complex genomic alterations that are challenging to detect with microarrays.
Improved Quantification and Data Quality
- RNA-Seq provides counts of reads mapping to each gene, offering a more direct and quantitative measure of gene expression compared to the relative fluorescence intensities of microarrays.
- The sequencing-based approach of RNA-Seq reduces background noise, enhancing the signal-to-noise ratio and improving the reliability of gene expression measurements.
Scalability and Technological Advancements
- Advancements in sequencing technologies have continually increased the throughput and reduced the cost of RNA-Seq, making it increasingly accessible for large-scale studies.
- RNA-Seq data can be seamlessly integrated with other next-generation sequencing data types (e.g., DNA sequencing, ChIP-Seq), facilitating comprehensive multi-omics analyses.
Better Handling of GC Bias and Other Technical Variations
- RNA-Seq protocols and bioinformatics tools have improved methods to account for GC content bias and other technical variations, leading to more uniform coverage across transcripts compared to microarrays.
Practical Implications of Choosing RNA-Seq Over Microarrays
The superior advantages of RNA-Seq translate into several practical benefits for researchers:
- Enhanced Discovery Potential: The ability to uncover novel transcripts and splice variants opens new avenues for understanding gene function and regulation.
- Precision Medicine Applications: Accurate detection of gene expression changes and genetic variants supports the development of personalized therapeutic strategies.
- Dynamic Range for Diverse Applications: From studying highly expressed housekeeping genes to rare transcripts involved in specific biological processes, RNA-Seq accommodates a wide spectrum of expression levels.
- Future-Proofing Research: As genomic annotations evolve, RNA-Seq data remains relevant and can be reanalyzed with updated references, whereas microarray data is constrained by the fixed probe sets used during the experiment.
Considerations and Challenges
Despite its numerous advantages, RNA-Seq also presents certain challenges:
-
Data Analysis Complexity: The vast amount of data generated by RNA-Seq requires robust computational infrastructure and expertise in bioinformatics for processing and analysis.
-
Higher Initial Costs: Although sequencing costs have decreased, RNA-Seq can still be more expensive than microarrays, especially for very large studies.
-
Standardization Issues: Variability in library preparation methods and sequencing platforms can introduce inconsistencies, necessitating standardized protocols for comparative studies.
Q03¶
What is the RNA Integrity Number (RIN)? How is it measured and why is it important in RNA-seq experiments? Describe what a low RIN indicates about a sample.
Solution
The RNA Integrity Number (RIN) is a standardized numerical value ranging from 1 to 10 that quantifies the integrity of RNA within a biological sample. Developed by Agilent Technologies, the RIN provides an objective measure of RNA quality, with higher values indicating intact RNA and lower values signifying degraded RNA. The RIN facilitates the comparison of RNA quality across different samples and experiments, enabling researchers to make informed decisions about the suitability of RNA for downstream applications like RNA-seq.
How is RIN Measured?
RIN is determined using specialized instrumentation, such as the Agilent Bioanalyzer or similar systems that perform capillary electrophoresis. The measurement process involves several key steps:
- Sample Preparation:
- RNA Extraction: RNA is first extracted from the biological sample using appropriate extraction methods to ensure purity and integrity.
- Quantification: The concentration of RNA is quantified, often using spectrophotometric methods, to ensure that sufficient material is available for analysis.
- Capillary Electrophoresis:
- Loading the Sample: A small volume of the RNA sample is loaded onto a microfluidic chip designed for RNA analysis.
- Separation of RNA Species: The RNA molecules are separated based on size through capillary electrophoresis. This process distinguishes between intact ribosomal RNA (rRNA) subunits and degraded RNA fragments.
- Automated Analysis:
- Electropherogram Generation: The instrument generates an electropherogram, a graphical representation of RNA fragments separated by size.
- Algorithmic Assessment: An integrated algorithm analyzes the electropherogram, evaluating various parameters such as the ratio of rRNA subunits (e.g., 28S to 18S in eukaryotic cells), the presence of degradation products, and the overall RNA distribution.
- RIN Calculation: Based on these assessments, the algorithm assigns a RIN value between 1 and 10, with 10 representing highly intact RNA.
Why is RIN Important in RNA-Seq Experiments?
RNA-seq is a powerful technique for profiling gene expression, identifying novel transcripts, and studying the transcriptomic landscape of cells and tissues. The accuracy and reliability of RNA-seq data are heavily dependent on the quality of the input RNA. Here's why RIN plays a pivotal role:
- Ensuring Data Reliability: High-quality RNA ensures that the sequencing data accurately reflects the true transcriptomic state of the sample. Degraded RNA can lead to incomplete or biased representation of transcripts.
- Reducing Technical Variability: By assessing RIN, researchers can standardize RNA quality across different samples, minimizing technical variability that could confound biological interpretations.
- Optimizing Library Preparation: RNA integrity affects the efficiency of library preparation steps such as fragmentation, reverse transcription, and adapter ligation. High-quality RNA facilitates more uniform and efficient library construction, leading to better sequencing outcomes.
- Enhancing Downstream Analyses: High RIN values contribute to more straightforward and accurate bioinformatics analyses, including alignment, quantification, and differential expression studies. Degraded RNA can introduce artifacts and complicate data interpretation.
- Resource Optimization: Ensuring high RNA quality before proceeding to expensive sequencing steps prevents the wastage of resources on suboptimal samples that may yield unusable data.
What Does a Low RIN Indicate About a Sample?
A low RIN value signifies that the RNA sample is degraded, meaning that the RNA molecules have been fragmented or broken down. Several factors can contribute to RNA degradation, including improper sample handling, delayed extraction, enzymatic activity (e.g., RNases), and suboptimal storage conditions. The implications of a low RIN in RNA-seq experiments are significant:
- Incomplete Transcript Representation: Degraded RNA often results in a bias towards the 3' ends of transcripts, as the 5' regions are more susceptible to degradation. This can distort gene expression profiles and affect the detection of full-length transcripts.
- Reduced Sequencing Efficiency: Fragmented RNA may lead to libraries with lower complexity, increasing the likelihood of redundant reads and reducing the ability to detect rare transcripts.
- Inaccurate Quantification: Degradation can cause uneven coverage across transcripts, leading to inaccurate quantification of gene expression levels and potentially misleading biological conclusions.
- Challenges in Bioinformatics Analysis: Fragmented reads may align ambiguously or incorrectly to the reference genome, complicating downstream analyses such as splice variant identification and differential expression.
- Increased Technical Noise: Degraded samples introduce technical noise that can obscure true biological signals, making it harder to discern meaningful patterns in the data.
- Potential Exclusion from Analysis: Samples with RIN values below a certain threshold may be excluded from RNA-seq analyses to maintain data integrity, potentially reducing sample size and statistical power.
Best Practices for Managing RIN in RNA-Seq Experiments
To mitigate the adverse effects of low RNA integrity, researchers should adhere to best practices during sample collection, handling, and processing:
- Rapid Sample Preservation: Promptly freezing samples or using RNA stabilization reagents immediately after collection can prevent RNA degradation.
- Minimizing RNase Exposure: Employing RNase-free consumables and maintaining a clean workspace reduces the risk of enzymatic degradation.
- Optimized RNA Extraction Protocols: Utilizing protocols that swiftly and effectively extract RNA while minimizing exposure to degradative conditions is essential.
- Proper Storage Conditions: Storing RNA samples at -80°C or lower preserves integrity over extended periods.
- Routine Quality Assessment: Routinely assessing RNA quality using RIN measurements allows for early detection of degradation and timely intervention.
- Sample Selection: Implementing quality thresholds based on RIN values ensures that only high-integrity RNA samples proceed to sequencing.
Q04¶
Describe the process of mRNA enrichment in RNA-seq experiments. Why is this step important, and how does it affect the resulting data?
Solution
Messenger RNA (mRNA) enrichment is a critical preparatory step in RNA sequencing (RNA-seq) experiments aimed at selectively isolating and concentrating mRNA molecules from a complex mixture of RNA species present within a biological sample. This process ensures that the subsequent sequencing effort is focused primarily on the transcripts that carry genetic information from DNA to the protein synthesis machinery, thereby enhancing the quality and relevance of the resulting data.
The Process of mRNA Enrichment
- Isolation of Total RNA: The first step in RNA-seq involves extracting total RNA from the biological sample, which includes a diverse array of RNA molecules such as ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), and various non-coding RNAs, in addition to mRNA. However, mRNA typically constitutes only about 1-5% of the total RNA in eukaryotic cells, making its isolation essential for efficient sequencing.
- Removal of rRNA:
Given that rRNA represents the majority of total RNA, its removal is paramount.
This can be achieved through methods such as poly-A selection or rRNA depletion.
- Poly-A Selection: Most eukaryotic mRNAs possess a polyadenylated (poly-A) tail at their 3' end. Poly-A selection leverages this feature by using oligo-dT (thymidine) primers attached to magnetic beads or other solid supports to hybridize and capture poly-A-tailed mRNAs. The non-polyadenylated RNAs, including rRNA and tRNA, remain unbound and are subsequently washed away, enriching the sample for mRNA.
- rRNA Depletion: Alternatively, specific probes can be designed to bind rRNA molecules, allowing their removal from the total RNA pool. This method is particularly useful for organisms or cell types where mRNA does not have a poly-A tail or for studies aiming to capture non-polyadenylated transcripts.
- Purification and Quality Control: After enrichment, the mRNA is purified to remove any remaining impurities and is then assessed for quality and quantity. Techniques such as agarose gel electrophoresis, Bioanalyzer profiles, or qPCR may be employed to ensure that the enrichment process has been successful and that the RNA is intact and free from degradation.
- Library Preparation: The purified mRNA is then converted into a sequencing library through processes like fragmentation, reverse transcription into complementary DNA (cDNA), adapter ligation, and amplification. This library is subsequently sequenced using high-throughput sequencing platforms to generate the data for analysis.
Importance of mRNA Enrichment
- Enhancing Data Quality and Efficiency: mRNA enrichment is crucial because it significantly increases the proportion of informative sequences in the RNA-seq data. Without enrichment, the sequencing reads would be overwhelmingly dominated by rRNA and other non-coding RNAs, leading to wasted sequencing resources and reduced coverage of mRNA transcripts of interest.
- Focusing on Coding Regions: By isolating mRNA, researchers can focus their analysis on the coding regions of the genome, which are directly involved in protein synthesis. This focus is essential for studies aimed at understanding gene expression patterns, identifying differentially expressed genes, and uncovering functional elements within the transcriptome.
- Reducing Sequencing Costs: Enriching for mRNA reduces the amount of sequencing required to achieve sufficient coverage of the transcripts of interest. This cost-effectiveness is particularly important for large-scale studies or when working with limited sequencing budgets.
- Improving Downstream Analysis: High-quality, mRNA-enriched samples facilitate more accurate and reliable downstream analyses, including gene expression quantification, variant detection, and transcript assembly. The reduced complexity of the library minimizes noise and enhances the signal-to-noise ratio, leading to more robust and interpretable results.
Impact on Resulting Data
- Increased Depth of Coverage: mRNA enrichment ensures that a larger proportion of sequencing reads correspond to mRNA molecules, thereby increasing the depth of coverage for each transcript. This enhanced coverage improves the ability to detect low-abundance transcripts and subtle changes in gene expression.
- Enhanced Detection of Differential Expression: With more reads mapping to mRNA, statistical analyses for differential gene expression become more powerful and sensitive. This increased sensitivity allows for the identification of genes that may play critical roles in biological processes or disease states but are expressed at lower levels.
- Facilitation of Transcriptome Complexity: mRNA enrichment aids in capturing the full complexity of the transcriptome, including alternative splicing variants and isoforms. This comprehensive representation is vital for understanding the diverse functional roles of genes and their regulatory mechanisms.
- Reduction of Bias and Artifacts: By minimizing the presence of highly abundant non-coding RNAs like rRNA, mRNA enrichment reduces potential biases and artifacts in the sequencing data. This leads to a more accurate reflection of the true mRNA landscape within the sample.
- Compatibility with Downstream Technologies: Enriched mRNA samples are better suited for integration with various downstream technologies and analyses, such as single-cell RNA-seq, where the limited amount of RNA requires efficient capture and representation of the transcriptome.
Q05¶
Explain why adding the $ symbol at the end of the string is important when constructing suffix arrays and Burrows-Wheeler Transform.
Solution
Adding the $ symbol (or any distinct end-of-string marker) at the end of a string is a crucial step when constructing suffix arrays and the Burrows-Wheeler Transform (BWT).
Without the $ symbol, constructing accurate and efficient suffix arrays and performing the BWT would be problematic, leading to ambiguities and potential errors in applications that depend on these techniques.
This special character plays several important roles that ensure the correct and efficient functioning of these data structures and algorithms.
Unique Termination of the String
- Avoiding Ambiguity: In the context of suffix arrays and BWT, suffixes of a string are sorted lexicographically. Without a unique termination symbol like
$, one suffix could be a prefix of another, leading to ambiguities in ordering. For example, consider the string"banana":- Suffixes:
"banana","anana","nana","ana","na","a" - Without
$, the suffix"a"is a prefix of"ana","anana", and"banana", making it unclear how to order them uniquely.
- Suffixes:
- Defining End Boundaries: The
$symbol clearly marks the end of the string, ensuring that every suffix is distinct. For example:- With
$:"banana$","anana$","nana$","ana$","na$","a$","$" - Now, each suffix has a distinct end, eliminating any ambiguity in their ordering.
- With
Ensuring Lexicographical Order
- Consistent Ordering: The
$symbol is typically defined to be lexicographically smaller than any other character in the string. This ensures that the suffix starting with$(i.e., the$itself) will always appear first in the sorted order of suffixes. This consistent ordering is essential for algorithms that rely on the first and last elements, such as BWT. - Boundary Conditions: In BWT, the first and last columns of the transformed matrix have special significance. The presence of
$ensures that these boundary conditions are well-defined and that the transformation can be reversed correctly.
Facilitating Reversal and Decoding
- Uniqueness for Reversal: The BWT is a reversible transformation, meaning you can reconstruct the original string from its BWT.
The
$symbol plays a vital role in this reversal process by providing a unique starting point. Without a unique end marker, it would be challenging to determine where the original string starts and ends. - Unambiguous Parsing: During the decoding process, the algorithm looks for the
$symbol to identify the end of the string. This unambiguous marker ensures that the reconstruction process can correctly determine the sequence of characters.
Optimizing Data Structures
- Efficient Sorting and Searching: In suffix arrays, having a unique termination symbol simplifies the sorting process of suffixes. It ensures that all suffixes are of different lengths and can be compared without running into issues where one suffix is a prefix of another.
- Simplifying Data Structures: The presence of
$allows certain optimizations in data structures like the suffix tree or the suffix array. It can reduce the complexity of algorithms by providing a clear stopping point.
Practical Applications and Standards
- Standard Practice: In many implementations and theoretical treatments of suffix arrays and BWT, appending a unique end-of-string symbol like
$is standard practice. This consistency aids in interoperability and understanding across different systems and implementations. - Error Prevention: Forgetting to append the
$symbol can lead to subtle bugs and incorrect behavior in applications that rely on suffix arrays or BWT, such as data compression algorithms (e.g., bzip2) and bioinformatics tools (e.g., genome sequence analysis).
Illustrative Example
Let's consider the string "banana" and see the difference with and without the $ symbol when constructing a suffix array and BWT.
Without $
Suffixes:
bananaananananaananaa
Sorted Suffixes:
aanaananabananananana
Issues: Ambiguity in ordering as "a" is a prefix of "ana", "anana", and "banana".
With $
- String:
banana$
Suffixes:
banana$anana$nana$ana$na$a$$
Sorted Suffixes:
$a$ana$anana$banana$na$nana$
Advantages:
- Each suffix is uniquely identifiable.
- Clear lexicographical order without ambiguity.
BWT Construction: The last characters of the sorted suffixes form the BWT: annb$aa
Q06¶
Create a suffix array for the string "mississippi$" and show your work. Present the suffix array as a list of starting indices.
Solution
First, let's list out the string with its corresponding 0-based indices:
| Index | Character |
|---|---|
| 0 | m |
| 1 | i |
| 2 | s |
| 3 | s |
| 4 | i |
| 5 | s |
| 6 | s |
| 7 | i |
| 8 | p |
| 9 | p |
| 10 | i |
| 11 | $ |
A suffix is a substring that starts at a particular index and extends to the end of the string.
Let's list all possible suffixes of mississippi$ along with their starting indices:
| Starting Index | Suffix |
|---|---|
| 0 | mississippi$ |
| 1 | ississippi$ |
| 2 | ssissippi$ |
| 3 | sissippi$ |
| 4 | issippi$ |
| 5 | ssippi$ |
| 6 | sippi$ |
| 7 | ippi$ |
| 8 | ppi$ |
| 9 | pi$ |
| 10 | i$ |
| 11 | $ |
Next, we sort the suffixes in lexicographical order. Here's how the sorting proceeds:
- $: The dollar sign is typically considered lexicographically smaller than any other character.
- i$: Starts with 'i'.
- ippi$: Starts with 'i', followed by 'p'.
- issippi$: Starts with 'i', followed by 's'.
- ississippi$: Starts with 'i', followed by 's', then another 's'.
- mississippi$: Starts with 'm'.
- pi$: Starts with 'p'.
- ppi$: Starts with 'p', followed by another 'p'.
- sippi$: Starts with 's', followed by 'i'.
- sissippi$: Starts with 's', followed by 'i'.
- ssippi$: Starts with 's', followed by 's'.
- ssissippi$: Starts with 's', followed by 's', followed by 'i'.
Here's the sorted list:
| Sorted Order | Starting Index | Suffix |
|---|---|---|
| 1 | 11 | $ |
| 2 | 10 | i$ |
| 3 | 7 | ippi$ |
| 4 | 4 | issippi$ |
| 5 | 1 | ississippi$ |
| 6 | 0 | mississippi$ |
| 7 | 9 | pi$ |
| 8 | 8 | ppi$ |
| 9 | 6 | sippi$ |
| 10 | 3 | sissippi$ |
| 11 | 5 | ssippi$ |
| 12 | 2 | ssissippi$ |
The suffix array is a list of the starting indices of the sorted suffixes.
Based on the sorted order above, the suffix array for "mississippi$" is:
Q07¶
Why is the BWT useful for compression? Explain with an example.
Solution
The Burrows-Wheeler Transform is instrumental in data compression due to its ability to:
- Enhance Data Redundancy: By rearranging the input to cluster similar characters.
- Improve Compression Efficiency: Making it easier for compression algorithms to reduce data size.
- Maintain Reversibility: Ensuring that the original data can be accurately retrieved.
BWT is a core component of compression tools like bzip2, which leverages BWT in conjunction with other compression techniques to achieve high compression ratios.
Purpose of the Burrows-Wheeler Transform (BWT)
- Rearrangement for Efficiency: BWT restructures the input data to enhance the performance of subsequent compression algorithms by increasing data locality and repeating patterns.
- Facilitation of Compression Algorithms: Transformed data exhibits more redundancy, which can be exploited by compression techniques like Run-Length Encoding (RLE) and Move-to-Front (MTF) encoding.
Analysis of Transformed String banana to annb$aa:
- Clustering of Identical Characters: Noticeable grouping of
as andns. - Enhanced Redundancy: Increased repetition aids compression algorithms.
Compression Techniques Leveraging BWT:
- Run-Length Encoding (RLE):
- Process: Encode consecutive repeated characters by their count.
- Example:
annb$aabecomesa1n2b1$1a2.
- Move-to-Front (MTF) Encoding:
- Process: Reorder characters based on their usage, placing recently used characters at the front.
- Benefit: Enhances the effectiveness of subsequent entropy encoding methods like Huffman coding.
Reversibility of BWT
- Key Property: BWT is reversible, allowing the original string to be perfectly reconstructed from the transformed data.
- Importance: Ensures data integrity during the compression and decompression processes.
Q08¶
Perform the Burrows-Wheeler Transform (BWT) for the following DNA sequence: ATTGCATTTGGCA.
Show your work (including the matrix) and specify what is the output.
Solution
To ensure that the BWT is reversible and to handle rotations correctly, we append a unique end-of-string marker, typically the $ symbol, which is lexicographically smaller than any other character.
- Original String:
ATTGCATTTGGCA - Modified String:
ATTGCATTTGGCA$
A rotation of a string is obtained by moving characters from the beginning to the end one by one.
For the string "ATTGCATTTGGCA$", which has 14 characters (indices 0 to 13), we'll generate all 14 rotations.
| Rotation | Rotated String |
|---|---|
| 0 | ATTGCATTTGGCA$ |
| 1 | TTGCATTTGGCA$A |
| 2 | TGCATTTGGCA$AT |
| 3 | GCATTTGGCA$ATT |
| 4 | CATTTGGCA$ATTG |
| 5 | ATTTGGCA$ATTGC |
| 6 | TTTGGCA$ATTGCA |
| 7 | TTGGCA$ATTGCAT |
| 8 | TGGCA$ATTGCATT |
| 9 | GGCA$ATTGCATTT |
| 10 | GCA$ATTGCATTTG |
| 11 | CA$ATTGCATTTGG |
| 12 | A$ATTGCATTTGGC |
| 13 | $ATTGCATTTGGCA |
Next, we sort all the rotated strings in lexicographical (dictionary) order. Here's the sorted list:
| Sorted Order | Rotation Index | Rotated String |
|---|---|---|
| 1 | 13 | $ATTGCATTTGGCA |
| 2 | 12 | A$ATTGCATTTGGC |
| 3 | 0 | ATTGCATTTGGCA$ |
| 4 | 5 | ATTTGGCA$ATTGC |
| 5 | 11 | CA$ATTGCATTTGG |
| 6 | 4 | CATTTGGCA$ATTG |
| 7 | 10 | GCA$ATTGCATTTG |
| 8 | 3 | GCATTTGGCA$ATT |
| 9 | 9 | GGCA$ATTGCATTT |
| 10 | 2 | TGCATTTGGCA$AT |
| 11 | 8 | TGGCA$ATTGCATT |
| 12 | 1 | TTGCATTTGGCA$A |
| 13 | 7 | TTGGCA$ATTGCAT |
| 14 | 6 | TTTGGCA$ATTGCA |
The Burrows-Wheeler Transform is obtained by taking the last character of each sorted rotation. Let's extract the last characters:
Final BWT Output: AC$CGGGTTTTATA
a) Without performing the full LF-mapping, explain how you would start searching for the pattern ATT in this BWT.
What would be your initial range in the F column, and what character would you look for first in the L column?
Solution
- Understanding F and L Columns:
- F Column: The first column of the BWT matrix, obtained by sorting the BWT string.
- L Column: The last column, which is the BWT string itself.
-
Constructing the F Column: Sort the BWT string
AC$CGGGTTTTATAlexicographically.- F Column:
$ A A A C C G G G T T T T T - L Column:
A C $ C G G G T T T T A T A
- F Column:
-
Initial Step for Pattern Matching ("ATT"): The pattern
"ATT"is processed from right to left, starting with the last character'T'. -
Determining the Initial Range in F Column:
- F Column:
$ A A A C C G G G T T T T T - Character to Find:
'T' - Positions of
'T'in F Column: Indices9to13(0-based indexing). - Initial Range:
[9, 13]
- F Column:
-
Character to Look for in L Column:
- First Character of the Pattern (Rightmost):
'T' - Looking for
'T'in the L Column within the initial range[9, 13]. - L Column:
AC$CGGGTTTTATA- Positions
9to13correspond to characters: L[9] = TL[10] = TL[11] = AL[12] = TL[13] = A
- Positions
- First Character to Look for in L Column:
'T'(the current character of the pattern being matched).
- First Character of the Pattern (Rightmost):
b) The sequence contains both TTT and TGC.
Without doing the full LF-mapping, explain which of these patterns would likely be easier to search for using the BWT, and why.
Solution
- Frequency and Distribution in BWT:
- Occurrences of
'T': Appears multiple times (Toccurs 5 times). - Occurrences of
'G'and'C':'G'appears 3 times;'C'appears 2 times.
- Occurrences of
- Pattern Characteristics:
- "TTT":
- Repeats the same character
'T'three times. - High frequency in BWT.
- Easier to locate due to consecutive
'T's.
- Repeats the same character
- "TGC":
- Sequence of distinct characters
'T','G','C'. - Less frequent as a specific sequence.
- Sequence of distinct characters
- "TTT":
- Ease of Search:
- "TTT":
- High occurrence increases the likelihood of multiple matches.
- Repeated characters simplify the search as transitions involve the same character.
- "TGC":
- Requires matching a specific sequence of distinct characters.
- Lower frequency and specific transitions make it harder to locate.
- "TTT":
"TTT" would likely be easier to search using the BWT because:
- The character
'T'is frequent and repeated. - The high occurrence of
'T'simplifies the matching process. - Repeated characters reduce the complexity of tracking transitions during the search.
c) How does the presence of unique substrings (like GCA) in the original sequence affect the structure of the BWT?
Explain why we see certain patterns in the BWT string.
Solution
- Unique Substrings:
- A substring like
"GCA"appears only once in the original string"ATTGCATTTGGCA$".
- A substring like
- Impact on Rotations and Sorting:
- Rotations Containing "GCA":
- The substring
"GCA"will appear in only one rotation. - This uniqueness affects the sorting of rotations, as the unique substring will anchor its specific rotation in a particular position within the sorted list.
- The substring
- Rotations Containing "GCA":
- Effect on BWT:
- Adjacent Characters:
- Characters preceding unique substrings in the sorted rotations influence the BWT.
- For example, if
"GCA"is preceded by a unique character in its rotation, the corresponding character in the BWT will reflect that.
- Patterns in BWT:
- Unique substrings lead to unique patterns or isolated characters in the BWT.
- Repeated substrings contribute to recurring patterns, while unique substrings introduce variability.
- Adjacent Characters:
- Illustrative Example with "GCA":
- Original Rotation Containing "GCA":
- Suppose one rotation is
GCA$ATTGCATTTGG(rotation index10).
- Suppose one rotation is
- Position in Sorted Rotations:
- This rotation's unique structure places it in a distinct position in the sorted list.
- Corresponding BWT Character:
- The character preceding
"GCA"in this rotation ('G') becomes part of the BWT.
- The character preceding
- Resulting in the BWT:
- This unique context ensures that the character
'G'appears in a specific position in the BWT, contributing to the overall structure.
- This unique context ensures that the character
- Original Rotation Containing "GCA":
- General Impact:
- Unique Substrings:
- Introduce unique characters or sequences in the BWT, making certain patterns stand out.
- Help in distinguishing specific contexts during pattern matching.
- Repetitive Substrings:
- Lead to recurring characters or patterns in the BWT, facilitating efficient compression and search operations.
- Unique Substrings:
Conclusion:
- Unique Substrings like
"GCA"create distinct patterns in the BWT string. - These unique sequences ensure that certain characters in the BWT are isolated or uniquely positioned, aiding in the differentiation of specific parts of the original string.
- The presence of unique substrings enhances the structural diversity of the BWT, which is beneficial for operations like pattern matching and data compression.
Q09¶
Reverse the Burrows-Wheeler Transform output BBAAB$AABA to get the original string.
What is the original string?
Show all of your steps.
Solution
The original string is ABBABAAAB$.
Programming+¶
These problems are not required and will not impact your BIOSC 1540 grade. The instructor will assess these separately to validate correctness without an assigned grade. Thus, you may work on these problems individually or in a team-based setting and "due" by the end of the semester. Happy coding!
Acceptable languages: Python v3.10+
Rewards
Engaging with these optional programming problems offers several valuable academic and professional growth opportunities.
- Consistent engagement with these Programming+ problems will allow me to write more detailed, compelling recommendation letters highlighting your computational skills. These personalized letters can significantly boost your applications for future academic programs, internships, or job opportunities.
- If there is enough interest, optional Friday recitations will be provided. This will give you individualized attention to accelerate learning and actionable feedback on your code and problem-solving approaches.
- Exceptional solutions may be featured on our course website with the students' permission. This is a way for us to recognize and appreciate your hard work and dedication to these problems.
Note
These problems would be similar to ones given in a major-only version of the class. Although, there would be more relevant instructions during class and would be given more than a week to complete.
P01¶
Implement a function that creates a suffix array for a given string.
Function Signature:
Requirements:
- The input string
swill not always end with a$character. - Use Python's built-in sorting function.
- Return a list of all suffixes of the input string, sorted lexicographically.
- Include the empty suffix (represented by
$) in the output. - Return the actual suffixes as strings, not their starting positions.
Example:
P02¶
Implement two functions: one to perform the Burrows-Wheeler Transform (BWT) and another to inverse the transform.
Function Signature:
Requirements:
- For
bwt(s):- The input string
swill not always end with a$character. - Use a simple approach, such as sorting all rotations of the string.
- Return the last column of the sorted rotation matrix as the BWT.
- The input string
- For
inverse_bwt(bwt):- The input
bwtis a BWT-transformed string. - Reconstruct and return the original string that produced the given BWT.
- The input
- Ensure that
inverse_bwt(bwt(s)) == sfor any valid inputs.
Example:
P03¶
Implement a function that performs pattern searching using a Burrows-Wheeler transformed string.
Function Signature:
Requirements:
- The
bwtinput is a Burrows-Wheeler transformed string. - Implement the backward search algorithm using the BWT.
- Return a list of integers representing the starting positions (0-indexed) of all occurrences of the pattern in the original text.
- Return an empty list if the pattern is not found.
- Correctly handle overlapping occurrences of the pattern.
- Implement necessary helper functions (e.g., for counting occurrences).
Example:
Hints:
- You may find it helpful to implement additional helper functions, such as:
- A function to compute the First column (F) from the Last column (L) of the BWT matrix.
- A function to perform the Last-to-First (LF) mapping.
- A function to count occurrences of characters in a prefix of the BWT string.
- Remember that the BWT string represents the last column of the sorted rotation matrix of the original string.
- The backward search algorithm involves iteratively narrowing down the range of rows in the BWT matrix that match the pattern, starting from the last character of the pattern.
A05
Be concise and focus on critical concepts. For each question or subpart, your response should be between 50 and 100 words.
Q01¶
Points: 8
Explain how X-ray crystallography is used to determine the electron density of a molecule. Describe how this information leads to fitting and refining an atomic model. Include the key steps in your explanation, from data collection to model refinement.
Solution
X-ray crystallography determines a molecule's electron density by directing X-rays at a crystallized sample. The X-rays scatter upon interacting with the electrons in the crystal, producing a diffraction pattern. By measuring the intensities and angles of these diffracted beams, scientists use Fourier transforms to calculate an electron density map, which reveals where electrons are likely to be located within the molecule.
This electron density information is crucial for fitting and refining an atomic model. Scientists fit atoms into regions of high electron density in the map, creating an initial model of the molecule's structure. They then refine this model by adjusting atomic positions and thermal factors to minimize discrepancies between the observed diffraction data and the model's predicted data.
Q02¶
Points: 8
Explain how X-rays scatter off electrons in a crystal. Discuss why this phenomenon is essential for determining protein structures using X-ray crystallography. In your answer, briefly describe the relationship between the crystal lattice and the resulting diffraction pattern.
Solution
X-rays scatter off electrons in a crystal through elastic interactions known as Thomson scattering. When X-rays encounter the electrons in a crystal lattice, they are deflected without a loss of energy. The electrons act as point scatterers, and due to the regular, repeating arrangement of atoms in the crystal, the scattered X-rays interfere constructively and destructively, creating a diffraction pattern.
This scattering phenomenon is essential for determining protein structures because the resulting diffraction pattern contains information about the electron density within the crystal. By analyzing the intensities and positions of the diffracted X-rays, researchers can compute an electron density map using Fourier transforms. This map reveals the locations of atoms in the protein, allowing scientists to build and refine an accurate three-dimensional model of its structure.
The relationship between the crystal lattice and the diffraction pattern is direct and fundamental. The periodic arrangement of atoms in the crystal lattice causes the scattered X-rays to interfere in specific directions, producing discrete spots in the diffraction pattern. According to Bragg's Law, the angles at which these spots appear are related to the spacing between lattice planes in the crystal. Thus, the geometry of the crystal lattice determines the positions and intensities of the diffraction spots observed.
Q03¶
Points: 8
Explain constructive and destructive interference in the context of X-ray diffraction patterns. Describe how these phenomena contribute to the formation of diffraction spots and the absence of certain reflections. Briefly discuss how understanding these concepts helps in interpreting X-ray diffraction data.
Solution
In X-ray diffraction, constructive and destructive interference arise from the superposition of X-rays scattered by electrons in a crystal lattice. Constructive interference occurs when the path difference between scattered waves leads them to be in phase, reinforcing each other and producing intense diffraction spots. Destructive interference happens when waves are out of phase, canceling each other out and resulting in the absence of reflections. These phenomena dictate the positions and intensities of diffraction spots, reflecting the crystal's internal structure. Understanding interference is crucial for interpreting diffraction data, as it allows scientists to calculate electron density maps and build accurate atomic models.
Q04¶
Points: 10
Compare and contrast Cryo-Electron Microscopy (Cryo-EM) and X-ray Crystallography as methods for determining the structures of biological macromolecules.
Solution
Cryo-Electron Microscopy (Cryo-EM) and X-ray Crystallography are techniques for determining macromolecular structures but differ in methodology and applications. X-ray crystallography requires crystallizing the sample, providing high-resolution structures but limited by the difficulty of crystallization for some proteins. Cryo-EM examines samples flash-frozen in vitreous ice without the need for crystals, capturing molecules in a more native state. Cryo-EM is advantageous for studying large complexes and membrane proteins, especially those difficult to crystallize. While X-ray crystallography often achieves higher resolution, Cryo-EM has improved significantly and is more suitable for heterogeneous or flexible samples.
Q05¶
a
Points: 4
Use UniProt to search for the Enoyl-[acyl-carrier-protein] reductase [NADH] (inhA) from Mycobacterium smegmatis Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) with the highest annotation score.
Summarize the function of this protein and provide the UniProt ID.
Change
I did not proof read enough and the species should have been Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv). I will accept structures from Mycobacterium smegmatis given the lateness of the correction.
Solution
UniProt ID: P9WGR1
The protein InhA is an enoyl-[acyl-carrier-protein] reductase in Mycobacterium tuberculosis, essential for mycolic acid biosynthesis, which is crucial for the bacterial cell wall. In the fatty acid elongation cycle, it catalyzes the NADH-dependent reduction of 2-trans-enoyl-[acyl-carrier protein]. This enzyme is a primary target of the tuberculosis drugs isoniazid and ethionamide, which inhibit its activity by forming an adduct with NAD. Mutations in inhA can lead to drug resistance, impacting tuberculosis treatment effectiveness.
b
Points: 2
Provide the amino acid sequence of the protein you found in part (a).
Solution
c
Points: 4
Visit the Protein Data Bank (PDB) and find the best experimental structure for native inhA and provide the four-character PDB ID.
Explain why you chose this structure (e.g., resolution, completeness, mutations).
Download the structure in PDBXmmCIF Format.
Tip
To select the best native structure of Enoyl-[acyl-carrier-protein] reductase (inhA) from the Protein Data Bank (PDB), follow these steps:
- Once the search results appear, you’ll see a list of structures associated with the protein. Each structure will have a unique PDB ID and may have different experimental conditions, resolutions, and details.
- To select the most suitable structure, consider the following:
- Structures with a lower resolution (in Ångstroms) are generally more accurate. Aim to select a structure with a resolution below 2.0 Å, as these offer higher detail. Structures with a resolution above 3.0 Å might be less precise.
- Check the structure for any missing regions, such as loops or domains. A complete structure provides a more accurate representation of the protein, while structures missing important regions may limit its reliability.
- Prefer structures solved using X-ray crystallography or Cryo-Electron Microscopy (Cryo-EM). X-ray crystallography typically yields high-resolution structures, but Cryo-EM can also provide important structural insights, especially for flexible regions.
- Review the structure’s description and any bound ligands. Choose a structure that reflects the biologically relevant state of the protein. For example, if studying the active form of the protein, select a structure where a relevant ligand or cofactor is bound.
- If available, examine the structure’s validation report, which provides insights into the accuracy and reliability of the structural data. Look for structures with good validation scores for quality assurance.
Q06¶
Points: 6
Describe the concept of homology modeling in protein structure prediction. Explain the main principles behind this method and why it's useful. Mention at least one limitation of this approach.
Solution
Homology modeling predicts a protein's three-dimensional structure based on its amino acid sequence similarity to a known structure (the template). The principle is that proteins with similar sequences tend to have similar structures. By aligning the target protein sequence with the template, a structural model is constructed by mapping residues accordingly. This method is useful for gaining structural insights when experimental data is unavailable, aiding in function prediction and drug design. A limitation is that the accuracy of the model heavily depends on the degree of similarity; low sequence identity between the target and template can result in unreliable models.
Q07¶
a
Points: 4
- Visit the SWISS-MODEL interactive workspace.
-
Use the following amino acid sequence for prediction:
-
After submitting the sequence, identify and record the five PDB IDs with the highest identity scores from the template search results.
- Build homology models using these templates.
- Explain what Global Model Quality Estimation (GMQE) is in the context of protein modeling.
- Note which PDB structure provided the highest GMQE score.
- Download the
modelCIFfile by clicking on the "Download Files" button associated with your best model.
b
Points: 2
- Navigate to AlphaFold 3 (AF3)
- Predict the structure of the inhA protein using the same amino acid sequence provided above.
- Once the prediction is complete, download and extract the results.
- Locate the file named
_model_0.cif, which contains the predicted structure from AF3.
c
Points: 4
- Load the following structures into Mol*.
You may use other programs such as PyMOL or ChimeraX.
(I personally prefer ChimeraX over PyMOL. I suggested Mol* since it is completely web based.)
- The experimental structure of the protein from Q05.
- The predicted structure from SWISS-MODEL (
modelCIFfile). - The predicted structure from AlphaFold 3 (
_model_0.ciffile).
- Align the structures to compare their conformations.
- Take a screenshot of the aligned structures and include it in your submission.
- Discuss at least one key difference observed between the structures. This could relate to structural features such as folding patterns, active sites, or any notable deviations.
Programming+¶
These problems are not required and will not impact your BIOSC 1540 grade. The instructor will assess these separately to validate correctness without an assigned grade. Thus, you may work on these problems individually or in a team-based setting and "due" by the end of the semester. Happy coding!
Acceptable languages: Python v3.10+, Mojo v24.4+, Zig v0.13+, Rust v1.80+
Rewards
Engaging with these optional programming problems offers several valuable academic and professional growth opportunities.
- Consistent engagement with these Programming+ problems will allow me to write more detailed, compelling recommendation letters highlighting your computational skills. These personalized letters can significantly boost your applications for future academic programs, internships, or job opportunities.
- If there is enough interest, optional Friday recitations will be provided. This will give you individualized attention to accelerate learning and actionable feedback on your code and problem-solving approaches.
- Exceptional solutions may be featured on our course website with the students' permission. This is a way for us to recognize and appreciate your hard work and dedication to these problems.
Note
These problems would be similar to ones given in a major-only version of the class. Although, there would be more relevant instructions during class and would be given more than a week to complete.
P01: PDB File Parser¶
In the field of structural biology, the Protein Data Bank (PDB) file format is widely used to store three-dimensional structural data of molecules. Your task is to create a program that can read and extract basic information from a PDB file. This will give you hands-on experience with the kind of data processing often required in structural biology research.
Write a program that takes a PDB file as input and extracts the following information:
- The total number of atoms in the structure
- The number of amino acid residues
To accomplish this, you'll need to understand the structure of a PDB file. Each line in a PDB file represents a record, and different record types contain different information. For example, ATOM records contain information about individual atoms, while HELIX and SHEET records describe secondary structures.
You may find it helpful to use the Biopython library, specifically the Bio.PDB module, which provides tools for working with PDB files. If you choose to use Biopython, look into the PDBParser class for reading PDB files and the Structure class for accessing the parsed data.
Your program should output a summary of the extracted information in a clear, readable format.
P02: RMSD Calculator¶
Root Mean Square Deviation (RMSD) is a commonly used measure to compare protein structures. It quantifies the average distance between atoms of superimposed proteins. This metric is crucial in various applications, such as assessing the quality of predicted protein structures or analyzing conformational changes.
Your task is to write a program that calculates the RMSD between two protein structures. The program should:
- Take two PDB files as input. These could represent the same protein in different states (e.g., experimental vs. predicted structures).
- Align the structures (you may assume they are already roughly aligned).
- Calculate the RMSD between corresponding atoms.
To calculate RMSD, you'll need to:
- Extract the 3D coordinates of corresponding atoms from both structures.
- Calculate the squared differences between these coordinates.
- Take the average of these squared differences.
- Take the square root of this average.
You may want to focus on comparing only the backbone atoms (N, Cα, C) or all heavy atoms. Be sure to specify in your output which atoms you're using for the comparison.
Consider using libraries like NumPy to handle the 3D coordinate data and perform calculations efficiently.
P03: Secondary Structure Predictor¶
Predicting protein secondary structure from amino acid sequence is a fundamental problem in bioinformatics. While modern methods use sophisticated machine learning techniques, simpler algorithms can still provide insights into the relationship between sequence and structure.
Your task is to implement a basic secondary structure prediction algorithm, such as the Chou-Fasman method. This algorithm assigns propensities for each amino acid to be in an alpha helix, beta sheet, or random coil, then uses these propensities to predict the most likely secondary structure for each residue.
Your program should:
- Take a protein sequence as input (a string of one-letter amino acid codes).
- Implement the Chou-Fasman algorithm or another simple prediction method.
- Output the predicted secondary structure for each residue (e.g., H for helix, E for sheet, C for coil).
To implement the Chou-Fasman method:
- Use a table of propensity values for each amino acid. You can find more information in the following papers: (Jiang et al., 1998 ; Chen et al., 2006)
- Scan the sequence to find regions with high propensities for helices or sheets.
- Apply rules for extending and terminating these structures.
- Assign remaining regions as coils.
Remember, this is a simplified method and won't be as accurate as modern predictors, but it will give you insight into the principles of sequence-based structure prediction.
P04: Solvent-Accessible Surface Area Calculator¶
Solvent-Accessible Surface Area (SASA) is an important property in protein structure analysis. It represents the surface area of a protein that is accessible to a solvent molecule (typically water) and can provide insights into protein-protein interactions, ligand binding sites, and more.
Your task is to write a program that calculates the SASA for each residue in a protein structure. The program should:
- Take a PDB file as input.
- Calculate the SASA for each residue.
- Output the SASA values in a clear, readable format.
To calculate SASA, you can:
- Use the Biopython library, which provides SASA calculation functionality.
- Use an external tool like FreeSASA, which you can call from your Python script.
- Implement a simple SASA algorithm yourself (this is more challenging and less accurate, but educational).
If using Biopython or FreeSASA:
- Read the documentation to understand how to use the SASA calculation functions.
- Ensure you understand the units of the output (typically Ų) and any parameters you need to set (like probe radius).
If implementing your own algorithm:
- Represent the protein as a set of spheres (atoms).
- Use a probe sphere to "roll" over the surface of these spheres.
- Calculate the area accessible to the probe.
Your output should list each residue along with its calculated SASA. Consider also providing summary statistics, like average SASA per residue type or total protein SASA.
A06
Be concise and focus on critical concepts. For each question or subpart, your response should be between 50 and 100 words.
Q01¶
Points: 8
Discuss how molecular dynamics (MD) simulations provide unique insights into protein behavior that static structural methods cannot capture.
Solution
Molecular dynamics (MD) simulations reveal dynamic properties of proteins by tracking atomistic movements over time, capturing both small-scale atomic vibrations and large-scale conformational changes. Unlike static structures obtained through methods like X-ray crystallography or cryo-electron microscopy, MD simulations provide time-resolved trajectories. This allows MD to capture protein flexibility, exploring a protein's conformational landscape, which is critical for understanding biological functions that rely on motion, such as binding, signaling, and catalysis.
Q02¶
Points: 8
In one sentence each, describe two key differences between molecular dynamics simulations and static structure determination methods.
Solution
- Molecular dynamics (MD) simulations capture the time-resolved motion of atoms, providing insight into the dynamic behavior of proteins that are missing from X-ray crystallography
- MD simulations can sample multiple conformations, revealing protein flexibility and various functional states, whereas static methods depict a single, often energy-minimized structure without showing alternate conformations.
Q03¶
Points: 5
Explain the approximations made when applying classical mechanics principles in molecular dynamics simulations.
Solution
In molecular dynamics (MD) simulations, classical mechanics approximations assume atoms are classical particles with defined positions and velocities, governed by Newton's laws of motion. This approach neglects quantum effects such as electron behavior, zero-point energy, and tunneling. Classical MD does not explicitly simulate electrons, instead using potential energy functions (force fields) to approximate their influence on atomic interactions. This approximation is useful for biomolecular systems where electronic excitations are not critical, but it limits MD's ability to model processes involving bond formation/breaking or reactions requiring quantum mechanics.
Q04¶
Points: 3
Identify a scenario in biomolecular simulations where quantum mechanical effects are significant, and explain why a classical MD approach might fail to accurately model this situation.
Solution
A scenario where quantum mechanical effects are significant is in the study of enzyme-catalyzed reactions, particularly those involving proton or electron transfer. These processes rely on quantum phenomena like tunneling and electronic transfers, which classical MD cannot capture, as it treats atoms as hard spheres with fixed charges and neglects electronic transitions. In this case, classical MD fails to model the reaction pathway accurately because it does not account for the wave-particle duality of electrons and the probability-based nature of quantum transitions
Q05¶
Points: 7
Briefly describe how Newton’s second law (\(F = ma\)) is utilized in MD simulations to simulate atomic motion.
Solution
In molecular dynamics (MD) simulations, Newton's second law (\(F = ma\)) is applied to compute atomic motion by calculating the acceleration of each atom based on the forces acting upon it. Using this relationship, the MD algorithm determines the acceleration (\(a\)) of each atom from the force (\(F\)) derived from the potential energy gradients of the system. This acceleration is then integrated over small time steps to update atomic velocities and positions, thus simulating the continuous motion of atoms over time.
Q06¶
Points: 5
Explain how the potential energy landscape influences the forces experienced by atoms during a simulation. Provide an example of a protein function that could be impacted by these force calculations.
Solution
In molecular dynamics simulations, the potential energy landscape defines the spatial distribution of energy based on atomic positions. Forces on atoms are calculated as the negative gradient of this potential energy, meaning atoms in regions of steep energy gradients experience stronger forces that drive them toward lower-energy conformations. For example, in enzyme active sites, the energy landscape guides substrate binding and positioning, which are crucial for catalytic function. Accurate force calculations help model the binding process and predict enzyme efficiency, as well as the influence of mutations on function.
Q07¶
Points: 3
Suppose you are simulating a protein bound to a novel small-molecule ligand. Explain the criteria you would use to select appropriate force fields for this system. Discuss the challenges associated with parameterizing the ligand if it is not represented in standard force field libraries, and outline the steps you would take to ensure accurate simulation results.
Solution
To select appropriate force fields for simulating a protein-ligand complex, I would prioritize compatibility with biomolecular systems, choosing a force field designed for proteins (e.g., AMBER or CHARMM) for the protein portion. For the ligand, which is novel, standard libraries may not provide parameters, creating challenges in accurately representing its unique bonds, angles, and non-covalent interactions. To address this, I would:
- Run QM calculations on the ligand to obtain reference data for its optimal geometry, charge distribution, and interaction energies.
- Use QM-derived data to develop custom force field parameters for the ligand, focusing on reproducing its electrostatic properties and bonded interactions within the protein environment.
- Perform preliminary MD simulations to check that the ligand maintains stability and realistic behavior, adjusting parameters as necessary by comparing with experimental or QM reference data.
This process ensures the ligand’s interactions are accurately modeled, providing reliable insights into the protein-ligand dynamics
Q08¶
Points: 3
Dihedral angles in molecular simulations often require periodic potential functions to capture rotational flexibility. Explain why Fourier series are used to model dihedral angle potentials, and describe how adjusting the terms in a Fourier series can influence the shape of the dihedral potential energy profile.
Solution
Fourier series are used to model dihedral angle potentials because they provide a flexible mathematical framework to describe periodic functions, which is essential for capturing the rotational nature of dihedral angles in molecules. The dihedral potential energy must repeat itself as the angle rotates, creating multiple stable conformations corresponding to energy minima. Fourier series achieve this by summing sine and cosine terms with varying frequencies to match the periodic energy profile.
Adjusting the terms in the Fourier series—such as the amplitude and frequency of each sine or cosine component—alters the shape of the dihedral potential. By modifying these terms, one can control the number and depth of energy minima and barriers, fine-tuning the energy landscape to reflect the specific conformational preferences and rotational flexibility of the dihedral angle in the molecule
Q09¶
Points: 7
Discuss how adding or removing specific ions or cofactors in a simulation could alter the observed protein behavior.
Solution
Adding or removing specific ions or cofactors in a simulation can significantly impact protein behavior by altering the protein's structural stability, dynamics, and function. Ions often stabilize certain protein conformations or are essential for activity in binding sites, where they help orient substrates or stabilize reaction intermediates. Removing these ions can disrupt critical interactions, leading to conformational shifts or loss of function. Similarly, cofactors (e.g., NADH in enzymes) are integral to catalytic activity, and their absence can prevent key functional states, altering the simulation outcome and potentially missing critical biological insights.
Q10¶
Points: 5
For a hypothetical protein simulation at pH 5, explain how this acidic environment could influence electrostatic interactions within the protein.
Solution
At pH 5, an acidic environment, certain amino acid side chains, such as aspartate (Asp) and glutamate (Glu), are more likely to be protonated, reducing their negative charge. This protonation affects electrostatic interactions within the protein by diminishing repulsive forces between negatively charged residues and altering salt bridge formations. Additionally, histidine (His), which has a pKa around 6, may become positively charged, creating new electrostatic attractions that stabilize alternate conformations. These changes can impact protein folding, stability, and function by altering the overall charge distribution and interaction network within the protein.
Q11¶
Points: 5
Explain why periodic boundary conditions (PBC) are essential for simulating realistic environments. Describe what could happen to a small protein simulation if PBC were not applied.
Solution
Periodic boundary conditions (PBC) are essential in molecular dynamics simulations to mimic an infinite, continuous environment by replicating the simulation box in all directions. This avoids artificial edge effects by ensuring that atoms leaving one side of the box re-enter from the opposite side, preserving the density and interactions that a molecule would experience in a larger system. Without PBC, a small protein simulation would experience unphysical behavior, with solvent molecules drifting away or clustering at the edges, creating an unrealistic environment. The lack of surrounding molecules could lead to inaccurate forces on the protein, impacting its stability and conformational sampling.
Q12¶
Points: 3
You are conducting a temperature-sensitive protein study. Evaluate the suitability of the Berendsen thermostat for accurately maintaining temperature. Compare it to the Nosé-Hoover thermostat, focusing on ensemble accuracy and how each method would affect the temperature distribution.
Solution
The Berendsen thermostat adjusts the temperature by scaling particle velocities uniformly, quickly bringing the system to the target temperature. However, it does not produce a true canonical (NVT) ensemble because it lacks realistic temperature fluctuations, leading to an artificially narrow temperature distribution. This can be problematic in temperature-sensitive studies where accurate ensemble representation is critical.
In contrast, the Nosé-Hoover thermostat connects the system to a "fictitious heat bath" by scaling particle momenta, allowing natural temperature fluctuations and thereby achieving a true NVT ensemble. This thermostat maintains a more realistic temperature distribution, which is essential for studies requiring precise temperature control. It better replicates the natural variations a protein might experience in a real environment.
Q13¶
Points: 5
A protein simulation shows high root mean square fluctuations (RMSF) in two specific loop regions. Discuss what this might indicate about the protein’s function and how this flexibility might impact the protein’s interactions with ligands or other biomolecules.
Solution
High root mean square fluctuations (RMSF) in specific loop regions indicate that these areas are highly flexible, which may be functionally significant. Loop flexibility is often associated with roles in binding or catalysis, as flexible regions can adapt their conformation to interact with various ligands or facilitate access to active sites. This flexibility allows the protein to accommodate different binding partners, potentially enhancing specificity and affinity. However, high flexibility may also lead to transient conformational changes that impact stability, requiring careful consideration in ligand or biomolecule interactions to ensure proper binding orientation and function.
Q14¶
Points: 3
Explain how the potential of mean force (PMF) is computed in molecular simulations and how it can help identify energy barriers in protein conformational changes.
Solution
The potential of mean force (PMF) in molecular simulations is computed by mapping the free energy along a chosen reaction coordinate, such as the distance between atoms or a dihedral angle. This is often done using techniques like umbrella sampling, where simulations are performed at different points along the coordinate with applied biases, followed by removal of these biases to obtain the unbiased free energy profile. The resulting PMF curve reveals energy minima and barriers along the reaction pathway, indicating stable states and the energetic cost of transitioning between them. For proteins, PMF analysis helps identify energy barriers that must be overcome for conformational changes, providing insight into the feasibility and frequency of these transitions in biological contexts
Q15¶
Points: 5
Explain how choosing a time step of 2 femtoseconds (fs) versus 0.5 fs could affect the simulation results. Discuss the potential consequences of using too large a time step on bond vibrations and overall system stability, and describe a scenario where a smaller time step would be preferable.
Solution
Choosing a time step of 2 femtoseconds (fs) instead of 0.5 fs can significantly impact simulation accuracy and stability. A larger time step, like 2 fs, allows faster computation but risks missing high-frequency motions, such as bond vibrations, leading to inaccuracies in simulating atomic interactions. If the time step is too large, bond vibrations may not be properly resolved, causing artifacts like unrealistic bond stretching, which can destabilize the system and lead to simulation failure.
A smaller time step, such as 0.5 fs, would be preferable in scenarios requiring precise representation of rapid motions, like simulating systems with light atoms (e.g., hydrogen bonds in water) or high-energy interactions. This approach captures finer details and maintains stability, though at a higher computational cost
Q16¶
Points: 5
Discuss the importance of ensemble averages in obtaining reliable data about system properties. Explain how the number and length of simulations with different initial conditions can impact the accuracy of ensemble averages.
Solution
Ensemble averages are crucial for obtaining reliable data about system properties in molecular simulations, as they represent the average behavior over many possible microstates. This approach accounts for the stochastic nature of atomic motion, providing accurate predictions of macroscopic properties like temperature, pressure, and free energy.
The accuracy of ensemble averages improves with the number and length of simulations, especially when each simulation starts with different initial conditions (e.g., randomized velocities). Multiple, longer simulations increase the sampling of microstates, reducing statistical noise and capturing rare events, which are essential for accurate property estimation. Inadequate sampling, either from too few or too short simulations, can lead to biased or incomplete results, underrepresenting certain conformations and limiting the reliability of calculated averages.
A07
Be concise and focus on critical concepts. For each question or subpart, your response should be between 50 and 100 words.
Q01¶
Points: 6
Identifying the correct protein target is a crucial step in drug development.
a) Explain why this identification is so important.
Solution
The identification of the correct protein target is crucial because it directly determines a drug's therapeutic effectiveness and potential side effects. Proteins drive disease processes through specific molecular mechanisms - for instance, by becoming overactive or misfolded. A well-chosen target allows the drug to address the root cause of disease rather than just symptoms. Additionally, incorrect target selection can lead to drug failure in clinical trials, wasted resources, and unwanted interactions with other proteins (off-target effects), potentially causing adverse reactions. This is why computational methods for target validation are so vital.
b) List and describe two criteria that make a protein a suitable drug target.
Solution
- Druggability: The protein must have features that allow small molecules or biologics to bind effectively, such as well-defined binding pockets or accessible surface areas. For example, enzymes and cell surface receptors are often good targets, while proteins with flat surfaces are challenging.
- Disease Association: Strong evidence must link the protein's activity to the disease state. Ideally, modulating the protein should directly affect the disease pathway with minimal impact on normal physiological processes. This can be validated through genetic studies, pathway analysis, and disease models.
- Expression Profile: The target should be expressed primarily in relevant disease tissues/cells and minimally in other tissues to reduce side effects. For example, a cancer target should be highly expressed in tumor cells but not in healthy tissue.
- Biological Validation: Scientific evidence (like knockout studies or clinical data) should confirm that modulating the target affects disease outcomes. This validates that hitting the target will have therapeutic value.
- Safety Window: The protein's function should be non-essential for normal cell survival or have sufficient redundancy to allow therapeutic intervention without causing severe toxicity.
- Assayability: The protein's activity should be measurable through reliable assays, enabling drug screening and optimization. This is crucial for both computational and experimental drug development.
- Structural Information: Available crystal structures or reliable structural models help in rational drug design and understanding binding mechanisms.
Q02¶
Points: 6
Gibbs free energy plays a significant role in the binding of a protein to a ligand.
a) Describe this role.
Solution
Gibbs free energy (ΔG) determines the spontaneity and strength of protein-ligand binding. A negative ΔG indicates spontaneous binding, with more negative values indicating stronger binding affinity. This free energy change represents the overall energetic favorability of the binding interaction, combining both enthalpic and entropic contributions.
b) How do enthalpic and entropic contributions influence the binding affinity between a protein and a ligand?
Solution
Enthalpic contributions (ΔH) reflect the formation of specific interactions like hydrogen bonds and van der Waals forces between the protein and ligand. Entropic contributions (ΔS) involve changes in system disorder, including the release of water molecules from binding surfaces (favorable) and the loss of conformational freedom upon binding (unfavorable). The balance between these factors (ΔG = ΔH - TΔS) determines the overall binding strength.
Q03¶
Points: 6
Noncovalent interactions contribute to the binding enthalpy in protein-ligand complexes.
a) List and briefly describe four types of these interactions.
Solution
- Hydrogen Bonds: Directional interactions between H-bond donors and acceptors, providing strength and specificity.
- Van der Waals Forces: Weak, distance-dependent attractions between atoms due to temporary dipoles.
- Electrostatic (Ionic) Interactions: Strong attractions between opposite charges, like salt bridges between acidic and basic groups.
- Hydrophobic Interactions: Favorable association of nonpolar groups, driven by water exclusion from binding pocket.
- π-π Stacking: Interactions between aromatic rings, important for binding aromatic ligands.
- Halogen Bonds: Directional interactions between halogen atoms and electronegative atoms.
- Cation-π Interactions: Attraction between cations and electron-rich π systems of aromatic rings.
b) Explain why these interactions are essential for binding specificity.
Solution
These interactions are essential for binding specificity because they create a unique recognition pattern in the binding site. Their diverse nature, directionality, and strength allow proteins to form selective interactions with specific ligands. The precise spatial arrangement and complementarity of these interactions ensure that only molecules with matching chemical features can bind strongly, much like a lock and key. This specificity is crucial for biological function and drug selectivity.
Q04¶
Points: 6
Entropy influences the binding of a ligand to a protein.
a) Explain how entropy affects this process.
Solution
Entropy affects protein-ligand binding through multiple competing factors. When a ligand binds, there is an unfavorable entropy loss due to restricted movement (reduced rotational, translational, and conformational freedom) of both molecules. However, this is often offset by favorable entropy gains from the release of ordered water molecules from hydrophobic binding pockets and protein-ligand interfaces. The net entropic contribution (ΔS) to binding is the sum of these opposing effects and significantly influences the overall binding free energy (ΔG).
b) Discuss why considering entropy merely as "disorder" is an oversimplification in the context of protein-ligand binding.
Solution
Viewing entropy solely as "disorder" oversimplifies the complex thermodynamic landscape of protein-ligand binding. While disorder is one aspect, entropy encompasses multiple sophisticated phenomena: the reorganization of water networks, changes in protein dynamics and flexibility, alterations in hydrogen bonding patterns, and coupled folding-binding events. Additionally, entropy can be locally ordered yet globally favorable - for example, when the release of ordered water molecules from a binding pocket leads to increased entropy in the bulk solvent, despite creating a more ordered protein-ligand complex.
Q05¶
Points: 6
Binding pocket detection is important in molecular docking studies.
a) Describe the importance of detecting binding pockets.
Solution
Detecting binding pockets is crucial for molecular docking because these sites determine where and how ligands can interact with the protein. Accurate pocket detection helps identify potential drug binding sites, predict protein function, and understand molecular recognition mechanisms. It also increases docking efficiency by limiting the search space to biologically relevant regions, reducing computational costs, and improving the accuracy of binding pose predictions. Additionally, pocket characteristics inform drug design by revealing the size, shape, and chemical features needed for optimal ligand binding.
b) What are the different types of binding sites, and how do they differ from each other?
Solution
- Orthosteric Sites: The protein's natural (primary) binding site for endogenous ligands. These are typically well-defined and conserved.
- Allosteric Sites: Secondary binding locations that modulate protein function through conformational changes. Often less conserved than orthosteric sites.
- Cryptic Sites: Hidden pockets that become accessible only upon protein conformational changes or ligand binding.
Q06¶
Points: 6
There are differences between systematic and stochastic search algorithms used in molecular docking.
a) Explain these differences.
Solution
Systematic search algorithms explore all possible conformations and positions of a ligand in a methodical, exhaustive manner by incrementally varying each degree of freedom. They guarantee finding the global minimum energy pose but are deterministic. In contrast, stochastic algorithms use random sampling of the conformational space, guided by probability distributions and acceptance criteria (like in Monte Carlo methods). They generate different results in each run and don't guarantee finding the global minimum but can efficiently explore large conformational spaces.
b) Why is stochastic sampling often preferred for larger molecules in docking studies?
Solution
Stochastic sampling is preferred for larger molecules because the number of possible conformations increases exponentially with molecular size (conformational explosion). For example, a molecule with 10 rotatable bonds would require 3^10 = 59,049 evaluations in a systematic search with just 3 positions per bond. Stochastic methods can efficiently sample this vast space by focusing on energetically favorable regions, making the calculation computationally feasible while still finding good solutions within reasonable time frames.
Q07¶
Points: 6
Ligand pose optimization is a critical step in docking studies.
a) Define ligand pose optimization.
Solution
Ligand pose optimization is the process of refining the position, orientation, and conformation of a ligand within a protein's binding site to find the most energetically favorable binding mode. This involves making small adjustments to the ligand's degrees of freedom (rotational, translational, and torsional) to minimize the binding energy and maximize complementarity with the binding site. The process typically uses local optimization algorithms to find the nearest energy minimum from an initial pose.
b) Why is it critical for accurately predicting binding affinity?
Solution
Pose optimization is critical for binding affinity prediction because small changes in ligand positioning can dramatically affect calculated interaction energies. An improperly optimized pose may miss key interactions (like hydrogen bonds) or create artificial clashes, leading to inaccurate energy calculations. Precise optimization ensures that all potential favorable interactions are captured and steric conflicts are resolved, providing a more reliable foundation for scoring functions to estimate binding affinity. This accuracy is essential for ranking different ligands in virtual screening.
Q08¶
Points: 7
Discuss how data-driven approaches, such as machine learning, can improve the efficiency and accuracy of virtual screening in drug discovery.
Solution
Machine learning (ML) improves virtual screening in several key ways. First, ML models can predict drug-likeness and ADMET properties rapidly, filtering out unsuitable compounds before expensive docking calculations. Second, ML can learn complex patterns from existing protein-ligand binding data to develop more accurate scoring functions that better predict binding affinities. Third, deep learning models can identify promising binding poses by learning from crystal structures, reducing false positives. Additionally, ML can help optimize search algorithms by learning which conformational spaces are most likely to yield successful binding poses.
These approaches significantly reduce computational costs while improving hit rates compared to traditional methods alone. For example, deep learning methods can screen millions of compounds in minutes, while physics-based methods might take weeks. This acceleration enables screening of larger chemical spaces while maintaining or improving accuracy.
Q09¶
Points: 7
Scoring functions are essential in molecular docking.
a) Explain their purpose.
Solution
Scoring functions evaluate and rank different ligand poses and compounds in molecular docking by estimating binding affinity. They serve multiple purposes:
- Identifying the most likely binding pose during pose optimization,
- Ranking different ligands to prioritize compounds for experimental testing, and
- Providing approximate binding free energy predictions.
They must balance accuracy with computational efficiency to handle large compound libraries in virtual screening campaigns.
b) How do machine learning-based scoring functions differ from traditional physics-based methods? Provide examples of advantages and limitations of each approach.
Solution
Physics-based scoring functions use fundamental principles like electrostatics and van der Waals interactions to calculate binding energies. They are interpretable and transferable across different protein families but often oversimplify complex effects like entropy and water dynamics.
ML-based scoring functions learn from experimental binding data to capture implicit patterns. Advantages include:
- Better handling of complex phenomena like water displacement
- Ability to learn non-obvious patterns from large datasets
- Faster evaluation time
Limitations include:
- Risk of overfitting to training data
- Limited extrapolation to novel chemical spaces
- Less interpretability compared to physics-based methods
- Dependence on high-quality training data
For optimal results, modern docking often combines both approaches.
Q10¶
Points: 7
Alchemical simulations are used to estimate the free energy of binding between a protein and a ligand.
a) Describe how these simulations work.
Solution
Alchemical simulations calculate binding free energy by gradually disappearing a ligand (or into another) through a series of non-physical (alchemical) intermediate states. These transformations follow a thermodynamic cycle where ligands are "disappeared" in both bulk solvent and protein binding site. The process uses advanced sampling techniques like thermodynamic integration or free energy perturbation to compute the free energy difference between states. The total binding free energy difference is then calculated by comparing the energetic costs of these transformations.
b) Explain why these simulations are computationally expensive, and discuss scenarios where they might be preferred over simpler docking methods.
Solution
These simulations are computationally expensive because they:
- Require multiple intermediate states for smooth transitions
- Need extensive sampling at each state to ensure convergence
- Must account for full protein flexibility and explicit solvent
- Often require microseconds of total simulation time
They are preferred when:
- Highly accurate binding free energies are needed (lead optimization)
- Comparing similar compounds with subtle structural differences
- Understanding detailed thermodynamic contributions to binding
- Evaluating effects of specific chemical modifications in drug design
- Standard docking fails to distinguish between similar ligands
Q11¶
Points: 7
Thermodynamic integration is a concept used in alchemical simulations.
a) Explain this concept.
Solution
Thermodynamic integration (TI) calculates free energy differences by integrating the ensemble average of the derivative of the system's Hamiltonian with respect to a coupling parameter (λ) that transitions between two states. The parameter λ typically varies from 0 to 1, representing the initial and final states. The integration is performed over multiple λ windows, where each window samples configurations at a specific intermediate state. This approach provides a rigorous way to compute free energy differences between states that aren't directly connected.
b) How does thermodynamic integration contribute to estimating free energy differences between bound and unbound states in protein-ligand interactions?
Solution
In protein-ligand binding, TI contributes to free energy estimation by:
- Computing the energetic cost of "disappearing" the ligand in both bulk solvent and protein binding site
- Sampling multiple λ windows to ensure smooth transitions between states
- Capturing protein conformational changes and solvent reorganization effects
- Providing a complete thermodynamic pathway between bound and unbound states
The total binding free energy is then calculated as the difference between the free energy changes in bulk and bound states, following a thermodynamic cycle.
Ended: Assignments
Exams ↵
Exams¶
We will have two exams: bioinformatics and computational structural biology.
Bioinformatics Exam
2024 Fall Bioinformatics Exam (Key)
Grade statistics

Question statistics

Review guide¶
This guide covers the major themes of the exam, providing a broad framework for your review. Since the exam is open note, concentrate on developing a deep understanding of the major concepts and approaches, rather than memorizing specific facts.
- Different Sequencing Approaches: Be familiar with the types of sequencing technologies used in bioinformatics.
- Read Types: Understand the differences between sequencing reads (e.g., single-end and paired-end) and how they influence downstream analysis.
- General Workflow Components: Know the steps involved in typical sequencing workflows and common issues that arise.
- Basic Terminology: Ensure you understand key terms related to genome assembly.
- General Algorithms: Be aware of the types of algorithms used for assembling genomes.
- Assembly Evaluation: Know the kinds of metrics used to evaluate genome assemblies and why they are important.
- Process Overview: Understand what gene annotation entails and how computational tools are used to predict gene function.
- Gene Features: Be familiar with the typical elements of a gene that are annotated.
- Types of Alignment: Understand the key differences between global and local alignment methods.
- Gap Penalty Models: Be aware of how gaps are handled during sequence alignment and why different models exist.
- Multiple Sequence Alignment (MSA): Know what MSA is used for and how it helps in comparing sequences.
- RNA-seq Overview: Understand what transcriptomics involves, especially in the context of RNA sequencing.
- Single-cell vs. Bulk Data: Know the differences between these approaches and why you would use one over the other.
- Normalization: Be familiar with the idea of normalization in RNA-seq data analysis and why it’s necessary.
- Basic Mapping Concepts: Know how reads are mapped to reference genomes and the key challenges involved in this process.
- Suffix Arrays: Understand the role of suffix arrays in bioinformatics.
- Different Approaches: Understand the general types of alignment strategies.
- Transcript Quantification: Be aware of the methods used to quantify gene expression levels.
- Generative Models in RNA-seq: Understand the purpose of generative models.
- Expectation-Maximization (EM) Algorithm: Be familiar with the general purpose of the EM algorithm.
- Two-Phase Inference Process: Understand Salmon's distinction between the online and offline phases.
- Transcript-Fragment Assignment Matrix: Know the role of the assignment matrix in RNA-seq data analysis.
- Statistical Models: Know why certain statistical models are used to analyze gene expression data.
- Interpreting Results: Be familiar with general principles for interpreting the significance of changes in gene expression.
Past exams¶
These are relevant, past exams. Note that in the Spring 2024 semester we had two quizzes instead of one exam.
Computational Structural Biology Exam
Grade statistics
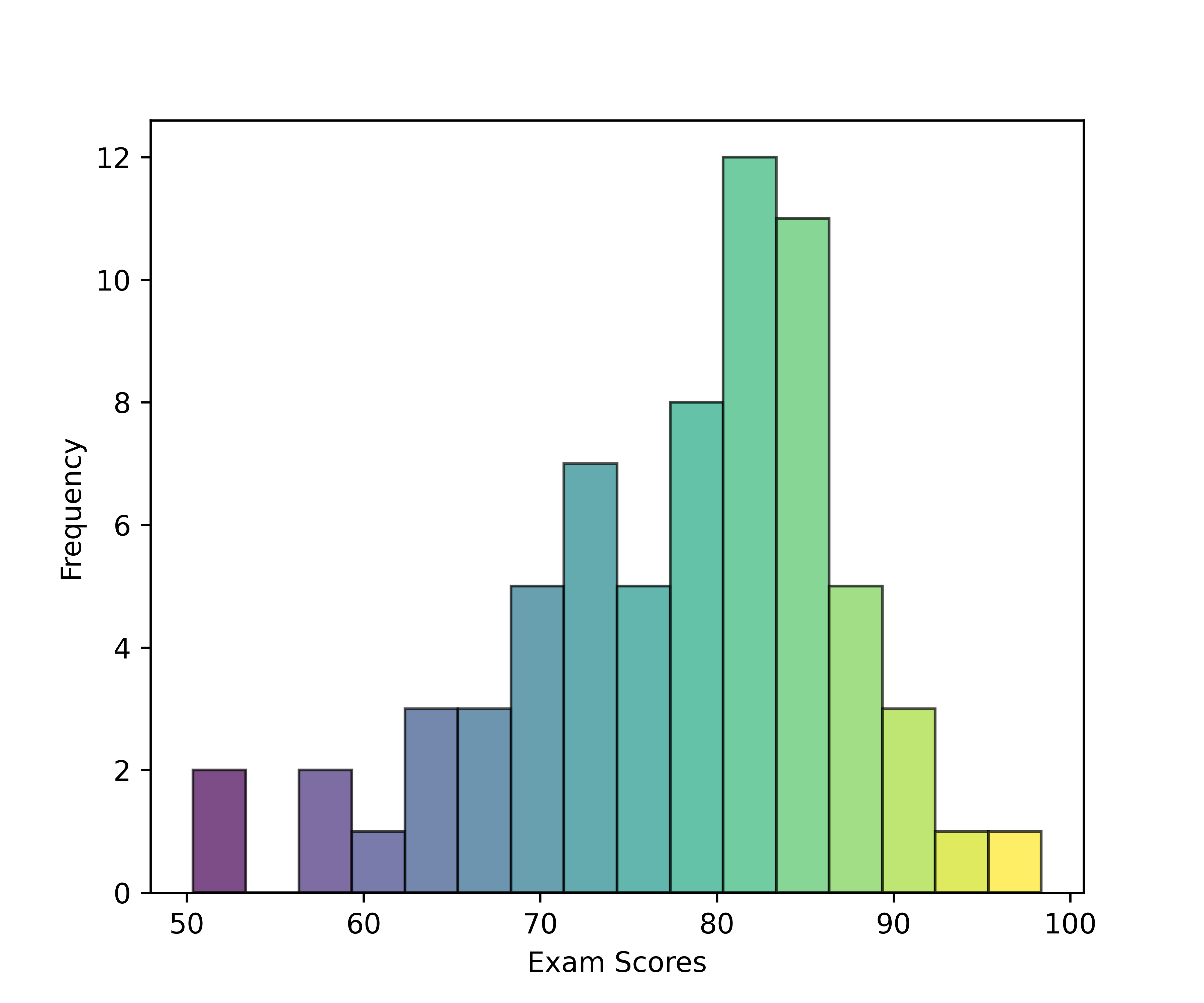
Question statistics
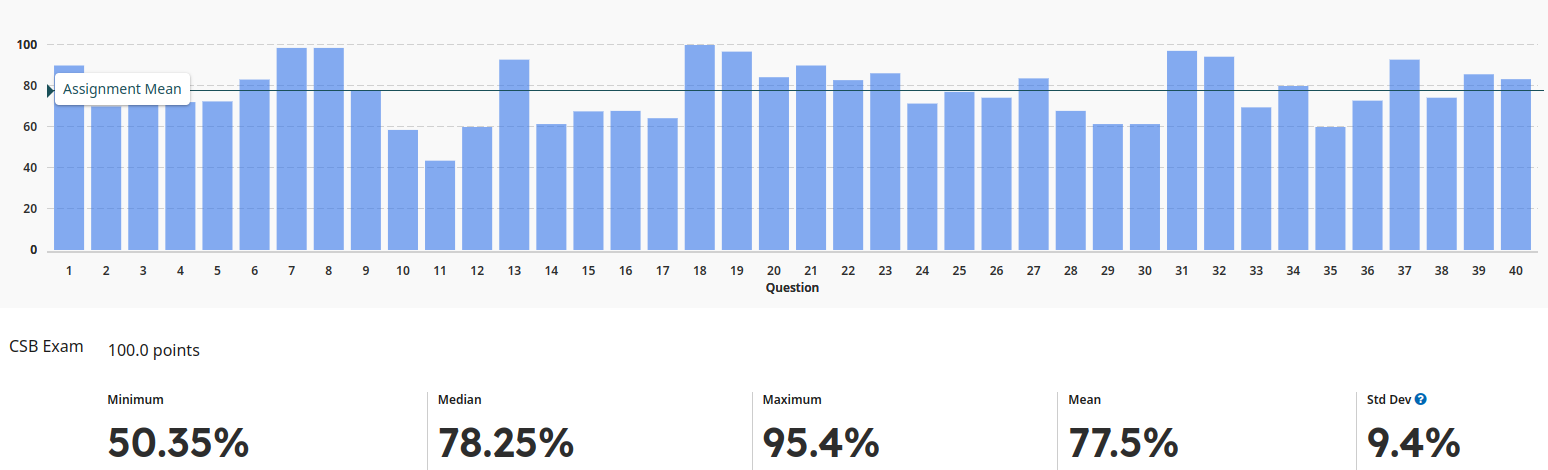
Review guide¶
This guide covers the major themes of the exam, providing a broad framework for your review. Since the exam is open note, concentrate on developing a deep understanding of the major concepts and approaches, rather than memorizing specific facts.
Lecture 11: Introduction to Structural Biology¶
- Explain the roles of covalent bonds, such as peptide bonds, in stabilizing primary protein structures, and how noncovalent forces drive protein folding and dynamic interactions.
- Describe the hierarchy of protein structure, including primary (sequence of amino acids), secondary (alpha helices, beta sheets), tertiary (3D shape of a polypeptide), and quaternary structures (multi-chain complexes).
- Understand how electron density maps from techniques like X-ray crystallography are used to determine atomic positions in protein structures.
- Explain how Cryo-Electron Microscopy (Cryo-EM) allows for protein structure determination without crystallization, and its importance in studying large, dynamic complexes in near-native states, especially for observing macromolecular assemblies.
- Explain the characteristics of intrinsically disordered proteins (IDPs), including their lack of stable 3D structure under physiological conditions and the challenges they pose for structure prediction compared to well-ordered proteins.
Lecture 12: Protein Structure Prediction¶
- Understand the importance of protein structure prediction in fields such as drug discovery, biotechnology, and the understanding of disease mechanisms.
- Explain the challenges in protein structure prediction, including Levinthal's paradox and the complexity of efficiently navigating conformational space.
- Describe the concept of rugged energy landscapes in protein folding and how proteins navigate these landscapes to achieve their functional conformations.
- Explain the basics of homology modeling, including the sequence similarity thresholds.
- Explain how coevolutionary analysis identifies pairs of residues that evolve together, indicating proximity in the 3D structure, and how this information informs structural predictions.
- Describe how AlphaFold uses deep learning, coevolutionary data, and neural network-based pattern recognition to predict protein structures with high accuracy.
Lecture 13: Molecular Dynamics (MD) Principles¶
- Explain the role of molecular dynamics (MD) simulations in studying protein folding, binding, and flexibility over time.
- Understand why atoms are modeled as classical particles in MD simulations and the implications of ignoring quantum mechanical effects.
- Apply Newton's laws of motion in the context of atomistic simulations within molecular dynamics.
- Describe the components of force fields, including bond stretching, angle bending, and torsions, and explain how they contribute to the potential energy in MD simulations.
- Identify scenarios in which quantum mechanical effects cannot be ignored and discuss the limitations of classical mechanics in these cases.
- Explain the key noncovalent interactions—hydrogen bonding, electrostatic interactions, van der Waals forces, and hydrophobic effects—and their roles in molecular recognition.
Lecture 14: Molecular System Representations¶
- Identify criteria for selecting high-quality experimental structures for simulations.
- Explain strategies for adding missing residues or atoms in protein models prior to conducting MD simulations.
- Describe the steps involved in protein preparation for simulations.
- Evaluate the suitability of a protein structure for simulations based on factors such as completeness, functional state, and clash scores.
- Explain the purpose of using periodic boundary conditions (PBC) in molecular simulations.
Lecture 15: Ensembles and Atomistic Insights¶
- Define microstates and macrostates, and explain their significance in statistical mechanics and molecular simulations.
- Discuss the importance of adequately sampling microstates in MD simulations to compute reliable ensemble averages.
- Explain how thermostats (like Berendsen and Nosé-Hoover) and barostats are used to control temperature and pressure in MD simulations.
- Distinguish between the equilibration (relaxation) phase and the production (data collection) phase in MD simulations.
- Use Root Mean Square Deviation (RMSD) as a metric to assess conformational changes in proteins over time during simulations.
- Use Root Mean Square Fluctuation (RMSF) to evaluate the flexibility of specific residues or regions in a protein during simulations.
- Explain the relationship between energy and probability in molecular simulations.
Lecture 16: Structure-Based Drug Design¶
- Describe the stages of the drug discovery pipeline and explain the role of computational methods.
- Identify key factors in selecting protein targets for drug design.
- Explain the role of virtual screening in narrowing down potential compounds from large chemical libraries during drug discovery.
- Describe how Gibbs free energy (ΔG) determines the strength of protein-ligand interactions.
- Explain the contributions of enthalpy (noncovalent interactions) and entropy (flexibility) to the thermodynamics of protein-ligand binding.
- Explain the purpose of alchemical free energy simulations in calculating changes in free energy.
- Describe how alchemical simulations work.
- Explain the method of thermodynamic integration (TI) used in alchemical simulations.
Lecture 17: Docking and Virtual Screening¶
- Describe the docking process in virtual screening, including how selecting one representative protein conformation simplifies protein-ligand binding prediction.
- Explain the importance of choosing an appropriate protein conformation to account.
- Describe methods for detecting binding pockets, such as alpha shape theory and grid-based techniques.
- Discuss the challenges in detecting cryptic binding sites.
- Explain the process of pose optimization in docking to optimize ligand positions within the binding site for accurate binding affinity prediction.
Lecture 18: Ligand-Based Drug Design¶
- Explain how molecular descriptors like LogP, molecular weight, and topological polar surface area (TPSA) are used to predict bioactivity in ligand-based drug design.
- Describe how Extended Connectivity Fingerprints (ECFPs) encode structural information for similarity comparisons in ligand-based drug design.
- Explain the basics of how molecular fingerprints are generated by hashing atom-specific properties.
- Discuss the challenges of efficiently exploring chemical space to find active compounds similar to known bioactive molecules.
Past exams¶
These are relevant, past exams.
- 2024 Spring CADD (No key was made)
Final Exam
This final exam was optional and allowed students to replace their lowest exam grade.
Grade statistics
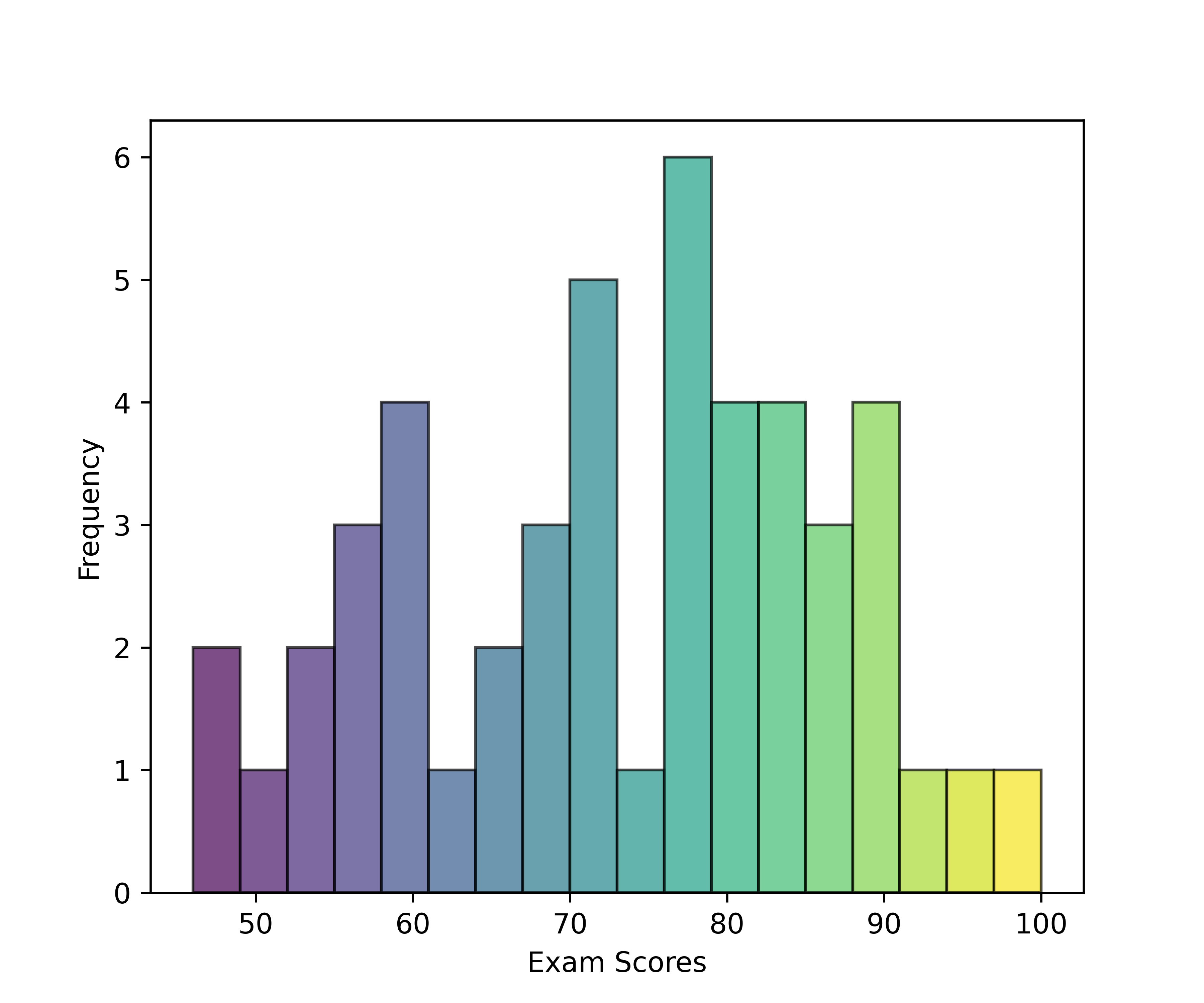
Question statistics
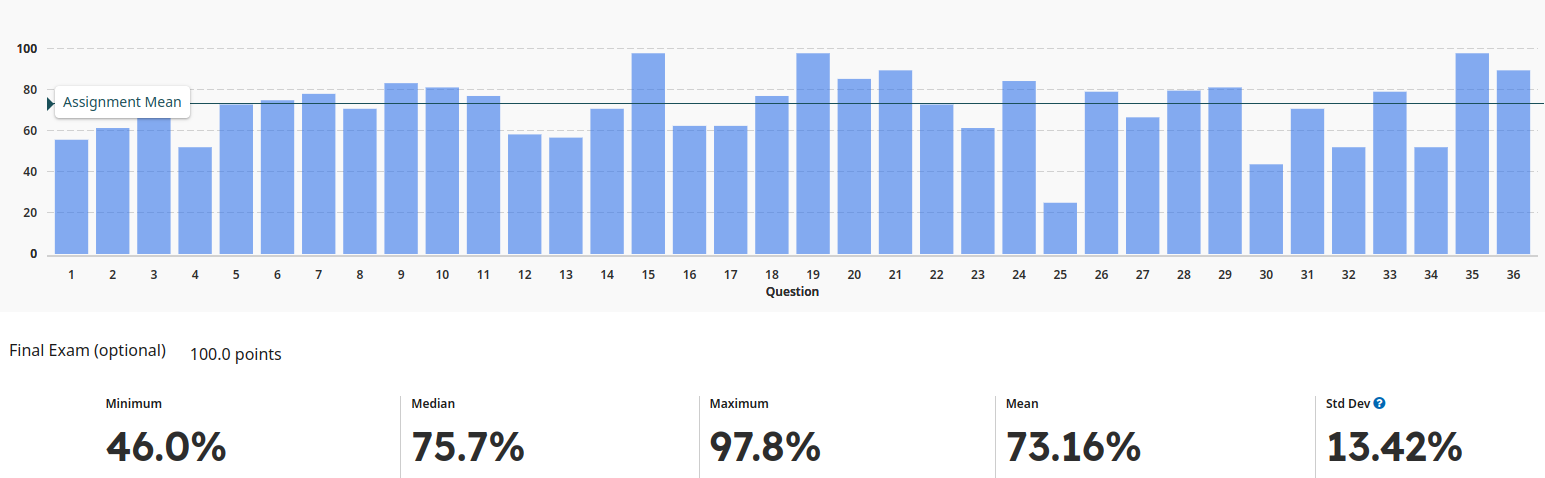
Ended: Exams
Project ↵
Project:
Computer-Aided Drug Design for a Novel Pathogen
This project aims to simulate the process of computer-aided drug design (CADD) for a novel pathogen, giving students hands-on experience with key bioinformatics and cheminformatics tools used in early-stage drug discovery. Students will work through a complete workflow—from raw sequencing data to potential drug candidates—mirroring the steps researchers might take when confronted with a new pathogenic threat.
Learning Objectives¶
By completing this project, students will:
- Gain practical experience with a complete CADD workflow.
- Understand how genomic data informs drug target selection.
- Learn to use and interpret results from state-of-the-art structure prediction tools.
- Develop skills in molecular docking and virtual screening.
- Critically analyze results at each stage of the drug discovery pipeline.
Overview¶
Genomic Data Processing and Analysis¶
This phase begins with raw Illumina sequencing reads (FASTQ format) from a provided SRA accession number 1. Students will perform quality control using tools like FastQC to ensure high-quality data for assembly. They will then assemble the pathogen's genome using appropriate assembly tools such as SPAdes. Following assembly, gene prediction and annotation will be done using tools like Prokka. Students will extract coding sequences, translate them to amino acid sequences, and perform functional annotation using tools such as InterProScan. This process will help identify potential drug targets based on function, essentiality, and other relevant criteria.
Students will use Galaxy.
Protein Structure Prediction¶
Students will use the amino acid sequences of selected target proteins to predict their 3D structures using AlphaFold3. This step introduces students to state-of-the-art protein structure prediction methods and the challenges of working with novel proteins. Students will learn to interpret AlphaFold's confidence metrics and structural output. They will also compare the predicted structures to known experimental structures in the Protein Data Bank (PDB) to assess the quality of the predictions and gain insights into the structural features of their targets. This comparison may involve structural alignment and analysis of key functional regions.
Students will use the AlphaFold web server.
Virtual Screening via Molecular Docking¶
In this final phase, students will screen a provided chemical library virtually against their predicted protein structures. This process begins with preparing the target protein structures for docking, including defining the binding site. Students will then set up and run the virtual screening, learning about the principles of molecular docking and scoring functions. After the screening, they will analyze the docking results to identify promising hit compounds. This analysis may include examining binding poses, interaction patterns, and docking scores. Students will learn to critically evaluate virtual screening results and understand their role in the early stages of drug discovery.
Students will use MolModa.
Deliverables¶
Students will compile and analyze their results from each stage throughout the project. Students will learn to integrate information from multiple computational approaches to make informed decisions about potential drug targets and hit compounds. They will prepare a final report summarizing their methods, results, and conclusions, demonstrating their understanding of the computer-aided drug design process.
- Assembled and annotated genome.
- Predicted 3D structures of selected target proteins.
- Docking results and analysis of top-hit compounds.
- Final report summarizing methods, results, and conclusions.
Part 1:
Genome assembly
You have been assigned a set of Illumina sequencing reads from a Staphylococcus aureus isolate. Your task is to assemble and annotate the genome, and then identify the dihydrofolate reductase gene, which is a common target for antibiotics. This project will give you experience with real-world bioinformatics tools and workflows used in genomics research.
Learning Objectives¶
By completing this project, you will:
- Gain practical experience with a complete genome assembly and annotation workflow.
- Learn to use and interpret results from key bioinformatics tools.
- Understand the process of identifying potential drug targets from genomic data.
- Develop skills in data analysis and interpretation in a genomics context.
Materials¶
You will be assigned one of 114 whole-genome sequencing runs from the BioProject PRJNA741582. Each run contains Illumina HiSeq 2000 paired-end reads of 250 nucleotides in length. Go here to find your SRA accession number.
Instructions¶
You will use a series of bioinformatics tools to process and analyze your assigned sequencing data. Follow these steps carefully, documenting your process and results at each stage.
A video walkthrough of the Galaxy portions of the project can be found here.
Setting Up Your Galaxy Project¶
Before beginning the analysis, you need to set up an account on Galaxy and create a project for this assignment. Galaxy is a web-based platform for data-intensive biomedical research that will allow you to perform all the necessary analyses for this project.
- Go to https://usegalaxy.org/
- Click on the "Login or Register" button in the top middle.
- If you don't have an account, click on "Register here" and fill out the registration form. Make sure to use a valid email address as you'll need to verify it.
- Once you've registered and logged in, click "Create new history+" and name your new project "BIOSC 1540 Project" and provide a brief description if you wish.
Click
Create.
Always ensure you're working within your "BIOSC 1540 Project" for this assignment. This will help you keep your work organized and easily accessible.
Galaxy will also save your work automatically, but it's a good practice to regularly check that your analyses are being saved correctly.
If you encounter any issues with Galaxy, check their support and training.
Getting data¶
In this step, you'll download the assigned sequencing data for your S. aureus isolate and upload it to Galaxy. Follow these instructions carefully:
- Your instructor will provide you with a unique SRA (Sequence Read Archive) accession number for your assigned S. aureus isolate.
We will use
SRX11246059as an example. - In the Galaxy interface, click on
Toolsin the left sidebar. - In the search bar at the top of the Tools panel, type
Download and Extract Reads in FASTQ format from NCBI SRAand click on it. - In the tool interface:
- For
select input type, chooseSRR accession. - In the
Accessionfield, enter your assigned SRA accession number. - Leave all other settings as default.
- For
- Click
Run Tool.
Galaxy will now download and extract your sequencing data. You can monitor the progress in the right sidebar, and will turn from blanched almond to green once finished.
Estimated time
7 minutes
- Once complete, you should see two new items in your history:
Single-end data (fastq-dump)Paired-end data (fastq-dump)
- There will be no data in
Single-end data (fastq-dump), so you can delete this. - To find the
forward.fastqcfile, you need to view job information forPaired-end data (fastq-dump), click on your accession number (i.e.,SRX11246059), then onforwardto preview its contents. You should see sequences in FASTQ format.
Report
Include the following information:
- Your SRA accession number that starts with
SRX. - The first five FASTQ entries in the
forward.fastqfile.
Quality Control with FastQC¶
Quality control is a crucial step in any sequencing data analysis. It helps you identify any issues with your sequencing data that might affect downstream analyses. We'll use FastQC, a widely used tool for quality control of high throughput sequencing data.
- In the Galaxy interface, search for "FastQC" in the tools panel.
- In the tool interface:
- For
Raw read data from your current history, selectDataset Collectionand choosePaired-end data (fastq-dump). - Leave all other settings as default.
- For
- Click
Run Tool.
Estimated time
1 minute
Once the job is complete, you'll see new items in your history for each FastQC report (Webpage and RawData).
Click on the FastQC on collection: Webpage, your accession number, and then both forward and reverse eye icons.
The report contains several modules, each assessing a different aspect of your sequence data.
Report
In your report, address the following questions for your forward reads:
- What is the overall quality of your sequencing data? Provide evidence from the FastQC report (i.e., screenshots) to support your assessment.
- Are there any concerning issues identified by FastQC? If so, what are they and how might they impact your downstream analyses?
- Based on the "Per base sequence quality" plot, is there evidence of quality degradation towards the end of the reads? If so, how might this inform your approach to read trimming?
- Do you see any evidence of adapter contamination or overrepresented sequences? If so, what steps might you take to address this?
- How do the results for your forward reads compare to your reverse reads? Are there any notable differences?
Remember, some warnings or failures in the FastQC report don't necessarily mean your data is unusable. The context of your experiment and the specific analyses you plan to perform should guide your interpretation of these results.
Adapter Trimming with Fastp¶
After assessing the quality of your raw sequencing data, the next step is to trim adapters and perform quality control. We'll use fastp, a fast all-in-one preprocessing tool for FASTQ files.
- In the Galaxy interface, search for "fastp" in the tools panel.
- Click on "fastp: fast all-in-one preprocessing for FASTQ files".
- In the tool interface, set the following parameters:
- "Single-end or paired reads": Select "Paired Collection".
- "Select paired collection(s)": Choose your paired-end reads.
- Click "Run Tool".
Estimated time
1 minute
Click on the eye icon next to the HTML report to view it. The report contains information about the trimming and filtering process.
Report
In your report, address the following questions with either your fastp or FastQC report:
- How many reads were removed during the filtering process? What percentage of reads passed the filters?
- Did fastp detect and trim any adapters? If so, which ones?
- How did the quality scores change after filtering? Provide specific examples from the report.
- Were there any issues with base composition (e.g., GC bias) before or after filtering?
- What was the duplication rate? Is this what you would expect for your type of sequencing data?
- Compare the fastp results to your earlier FastQC results. How do they complement each other? Are there any discrepancies?
Remember, the goal of this step is to improve the overall quality of your sequencing data by removing low-quality reads, trimming adapters, and addressing any other issues identified in the FastQC step. The fastp results should show an improvement in data quality compared to the raw reads.
De Novo Genome Assembly with SPAdes¶
After trimming and quality control, the next step is to assemble the processed reads into contigs. We'll use SPAdes, a versatile genome assembler designed for both small genomes and single-cell projects.
- In the Galaxy interface, search for "SPAdes" in the tools panel.
- Click on "SPAdes genome assembler for genomes of regular and single-cell projects".
- In the tool interface, set the following parameters:
- "Operation mode": Select "Assembly and error correction"
- "Single-end or paired-end short-reads": Choose "Paired-end: list of dataset pairs"
- "FASTA/FASTQ file(s)": Select your paired-end output from the fastp step
- Click "Run Tool".
Estimated time
2 hours
Once complete, you'll see new items in your history, including the assembly graph, contigs, and scaffolds.
To get more detailed statistics about your assembly, we'll use the Bandage Info tool:
- In the Galaxy interface, search for
Bandage Infoin the tools panel. - Click on
Bandage Info. - In the tool interface, set the following parameters:
Graphical Fragment Assembly: Select theSPAdes: Assembly Graph with Scaffolds.- Leave all other settings as default.
- Click "Run Tool".
- Once complete, you'll see a new item in your history with the Bandage Info output.
- Next, repeat the same process with
Bandage Imageand include this in your report.
Estimated time
1 minute
Report
In your report, address the following questions:
- How many contigs (i.e., nodes) were produced by the assembly?
- What is the total length of the assembly? How does this compare to the expected genome size of Staphylococcus aureus (approximately 2,800,000 base bp)? (Note: You can also use quast to check this number.)
- Calculate the difference between the "Total length" and "Total length no overlaps". What does this difference represent, and why is it important to consider?
- What is the N50 of your assembly? How does this value compare to the median node length? What does this tell you about the distribution of contig sizes in your assembly?
- Analyze the "dead ends" in your assembly. How many dead ends are there? What percentage of the total possible ends (2 * number of nodes) do these represent? What might a high percentage of dead ends indicate about your assembly?
- Based on all these results, how would you assess the overall quality of your genome assembly? Consider factors such as completeness, contiguity, and potential issues.
- How do you think the "careful" mode in SPAdes might have influenced these assembly statistics compared to the default mode?
Gene Annotation with Prokka¶
After assembling the genome, the next step is to annotate it to identify genes and their potential functions. We'll use Prokka, a rapid prokaryotic genome annotation tool.
- In the Galaxy interface, search for
Prokkain the tools panel. - Click on
Prokka: Prokaryotic genome annotation. - In the tool interface, set the following parameters:
- "Contigs to annotate": Select the Scaffolds file from your SPAdes output
- "Genus name": Enter "Staphylococcus"
- "Species name": Enter "aureus"
- Click "Run Tool".
Estimated time
10 minutes
After the annotation is complete, examine the output files. Pay particular attention to:
- GFF3 file (.gff): This contains the annotations in a standard format. You can use this file to visualize the annotations in a genome browser.
- GenBank file (.gbk): This file contains both the sequence and the annotations. It can be viewed in many sequence analysis tools.
- Statistics file (.txt): This provides a summary of the annotation process. Look at the number and types of features identified.
- Protein FASTA file (.faa): This contains the amino acid sequences of predicted proteins. You'll use this to find your gene of interest (dihydrofolate reductase).
Report
In your report, address the following questions:
- How many coding sequences (CDS) did Prokka identify in your genome?
- How many rRNA and tRNA genes were annotated?
- What other types of features did Prokka identify, and how many of each?
- How does the number of annotated genes compare to what you would expect for a Staphylococcus aureus genome?
- Search the
.faafile for the dihydrofolate reductase gene. Did Prokka identify this gene? If so, what is its protein ID? What is the amino acid sequence of your protein? - Based on these results, how would you assess the quality and completeness of your genome annotation?
- How might the genus and species names you provided influence the annotation process?
Remember, a typical S. aureus genome contains around 2,500 to 2,900 genes. The presence or absence of certain genes can provide insights into the strain's potential characteristics or capabilities.
Comparing to UniProt¶
After annotating your genome and identifying the dihydrofolate reductase gene, the next step is to compare your sequence to a known reference. This comparison will help you verify your annotation and identify any potential variations in your sequence.
- Go to the UniProt website (https://www.uniprot.org/).
- Find the dihydrofolate reductase protein (folA gene) entry for Staphylococcus aureus (Taxon ID
1280).
Report
In your report, address the following questions:
- What is the UniProt ID?
- What is the length of your protein sequence? How does it compare to the UniProt reference (159 amino acids)?
- Calculate the percentage identity between your sequence and the UniProt reference. Are they identical, or are there differences?
- If there are differences, list their positions and the amino acid changes. Are these changes conservative (similar amino acids) or non-conservative?
Submission Guidelines¶
- Submit your report as a single PDF file named
LastName_FirstName_GenomeAssemblyReport.pdfto Gradescope. - Responses to questions should be numbered under a heading and be of reasonable length.
- Ensure all figures and tables in your report are clearly labeled and referenced in the text.
Rubric¶
Your answers to all the aforementioned questions will help you garner points according to this rubric.
Data Acquisition and Initial Processing
| Criterion | Points | Description |
|---|---|---|
| SRA Data Retrieval | 10 | - Successfully downloads correct SRA dataset (5 pts) - Properly identifies and presents first five FASTQ entries (5 pts) |
Quality Control Analysis
| Criterion | Points | Description |
|---|---|---|
| FastQC Interpretation | 12 | - Accurately interprets quality metrics (4 pts) - Identifies key issues in sequencing data (4 pts) - Compares forward/reverse reads effectively (4 pts) |
| Fastp Analysis | 8 | - Correctly reports filtering statistics (3 pts) - Analyzes quality improvement after trimming (3 pts) - Interprets duplication rates appropriately (2 pts) |
Genome Assembly
| Criterion | Points | Description |
|---|---|---|
| Assembly Statistics | 15 | - Accurately reports and interprets N50 (4 pts) - Analyzes contig numbers and lengths (4 pts) - Compares assembly size to expected genome size (4 pts) - Evaluates assembly graph characteristics (3 pts) |
| Quality Assessment | 10 | - Critically evaluates assembly quality (5 pts) - Identifies potential issues and limitations (5 pts) |
Genome Annotation
| Criterion | Points | Description |
|---|---|---|
| Prokka Results | 15 | - Reports complete annotation statistics (5 pts) - Analyzes gene content and distribution (5 pts) - Compares results to expected S. aureus features (5 pts) |
| Gene Analysis | 10 | - Identifies dihydrofolate reductase gene (5 pts) - Correctly reports gene characteristics and location (5 pts) |
Protein Analysis and Comparison
| Criterion | Points | Description |
|---|---|---|
| UniProt Comparison | 10 | - Performs thorough sequence comparison (4 pts) - Identifies and analyzes sequence variations (3 pts) - Interprets biological significance of findings (3 pts) |
Report Quality
| Criterion | Points | Description |
|---|---|---|
| Scientific Writing | 6 | - Clear, concise scientific writing |
| Format Compliance | 2 | - Follows submission guidelines - Proper file naming - Complete PDF submission |
Part 2:
Protein structure prediction
In this phase, you will predict and analyze the structure of dihydrofolate reductase (DHFR) from S. aureus that you identified in Part 1. DHFR is an essential enzyme involved in folate metabolism and an important antibiotic target.
By completing this project, you will:
- Gain practical experience with state-of-the-art protein structure prediction tools.
- Learn to critically evaluate and compare protein structure predictions.
- Understand how to analyze protein structures and their functional features.
- Develop skills in structural visualization and analysis.
Instructions¶
Use PyMOL for your structural analysis. You often will need to use the "licorice" representation of the protein to see that atomistic differences.
To support your computational biology project, your instructor has provided a PSE file for each question. A PSE file is a PyMOL session file that contains a pre-configured molecular structure and relevant visualizations to help you address the specific questions. These files are designed to save you time and effort by providing the necessary setup, including loaded molecules, annotations, and possibly preset views that align with the problem requirements.
In each screenshot, ensure you provide a visual indication and/or label to specify the origin (e.g., I-TASSER or 6PRD) of the structure. Below is an example of an appropriate image to use in your report.
Example image
Conformations of His23, Glu143, and Lys144 of DHFR are shown below for 3FRD and 6PR6 with carbon atoms shown in pink and white, respectively.
Structures were aligned by their alpha carbons.
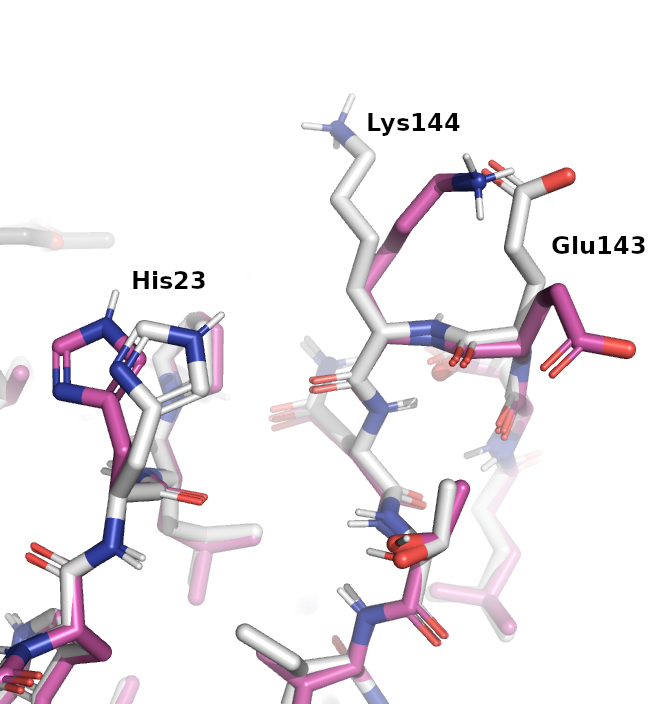
Structure predictions¶
Using the UniProt DHFR sequence from your genome assembly project, we will perform several predictions. Instead of flooding these free servers with the same jobs, we will all use the same outputs.
- I-TASSER (Job ID S799334): A threading-based method for predicting protein structures and functions by assembling models from template fragments and refining them iteratively; it is robust for cases with moderate to strong homologous templates and provides functional annotations, often generating medium-resolution models.
- D-I-TASSER (Job ID DIT6377): An advanced iteration of I-TASSER using deep-learning-based spatial restraints to model proteins with greater accuracy, especially in challenging cases with weak homology, providing more reliable multi-domain structure predictions compared to I-TASSER.
- I-TASSER-MTD (Job ID ITM552669806): Designed specifically for multi-domain proteins, it combines domain parsing, single-domain folding, and inter-domain assembly with improved functionality and accuracy for large, multi-domain proteins compared to D-I-TASSER
- SWISS-MODEL (Job ID a7BMLv): A homology modeling platform emphasizing user accessibility and high-quality models through evolutionary template matching; while not as suitable for low-homology cases as I-TASSER or AlphaFold3, it excels in providing intuitive results for well-conserved targets
- AlphaFold3 (Download the PDB file here): A state-of-the-art model leveraging a diffusion-based architecture to predict highly accurate protein structures and biomolecular interactions across diverse targets, outperforming specialized tools in protein-ligand and protein-nucleic acid interactions.
Submission
In your submission, answer the following questions:
- Download the PDB files for SWISS-MODELS 01 (6E4E), 05 (3FYW), 03 (6PRP), and 06 (6PR8). Identify if there are any apparent protein conformational differences near the NADP(H) and folate binding pockets. Provide screenshots to support your claims. (PyMol session file)
- Download the PDB file for SWISS-MODEL 02 (2W3M) and compare it against model 01 (6E4E). What is the alpha-carbon RMSD after alignment? Out of these two structures, which would you use for docking? Justify your choice. Provide screenshots to support your claims. (PyMol session file)
- Download the I-TASSER, D-I-TASSER, I-TASSER-MTD, and your AlphaFold3 PDB structures. Compare these structure to each other and to SWISS-MODEL 01. Which prediction has the highest similarity (i.e., low RMSD) to the SWISS-MODEL? Which method would you generally find more reliable? Provide screenshots to support your claims. (PyMol session file)
Experimental structures¶
The following experimental structures were selected for our analysis. All are wild-type S. aureus DHFR with co-crystallized NADP(H).
| PDB ID | Additional ligand |
|---|---|
| 3FRD | Folate |
| 6PRA | None |
| 6PRB | OWM |
| 6PRD | OWG |
| 6PR6 | OWS |
| 6PR7 | OWP |
| 6PR8 | OWJ |
| 6PR9 | OWV |
Remember that you need to add hydrogens to your structures!
Submission
In your submission, answer the following questions:
- Download 3FRD and 6PRA PDB files. Suppose you will be docking in the folate pocket while keeping NADP(H) and removing all water molecules. Are there any structural differences in the protein that could impact your docking results? If yes, specify which residues, explain how this would impact docking, and provide screenshots to support your claims. If no, provide evidence in the form of screenshots and a possible explanation. (PyMol session file)
- Identify noncovalent interactions in 3FRD that stabilize this folate pose. Use screenshots to support your claims. (Use the same PyMol session file as the previous question.)
- Download all PDB structures in the above table that start with
6PR. Each of these6PRstructures, excluding 6PRA, contain co-crystallized ligands that are potential S. aureus DHFR inhibitors from this publication. Identify protein-ligand noncovalent interactions for each inhibitor and note similarities and differences between folate noncovalent interactions. (PyMol session file) - In the same
6PRstructures, identify amino acids that are in different conformations depending on the ligand. Also, which protein would be the least reliable for subsequent protein-ligand docking? Provide a rationale. (Use the same PyMol session file as the previous question.)
Part 3:
Docking
In the ongoing battle against antibiotic-resistant bacteria, understanding the molecular interactions between drugs and their targets is crucial. Staphylococcus aureus dihydrofolate reductase (DHFR) is a well-established target for several antibiotics due to its essential role in bacterial folate synthesis. This project focuses on protein-ligand docking studies of S. aureus DHFR using MolModa, a powerful tool for virtual screening and molecular modeling.
You will perform docking simulations to predict how various ligands bind to DHFR within different protein environments. By comparing these predictions with experimental data, you will assess the reliability of computational docking as a method for identifying potential antibiotic candidates. Additionally, you will explore the impact of cofactors like NADPH on ligand binding, providing insights into the complexities of protein-ligand interactions in a cellular context.
Learning Objectives¶
By completing this project, you will:
- Comprehend the principles and significance of molecular docking in drug discovery.
- Navigate and utilize MolModa for preparing proteins and ligands for docking.
- Configure and execute docking simulations with appropriate parameters.
- Compare computational docking scores with experimental binding affinities (IC50, Ki, MIC).
- Assess the impact of cofactors, such as NADPH, on ligand binding and docking outcomes.
- Evaluate the correlation between docking predictions and experimental data.
- Generate and interpret visual representations of ligand poses and binding interactions.
- Analyze how decoys are scored and the implications for screening accuracy.
Instructions¶
We will be using MolModa to perform protein-ligand docking of S. aureus DHFR in the following proteins.
| PDB ID | Additional ligand |
|---|---|
| 3FRD | Folate |
| 6PRA | None |
| 6PR6 | OWS |
MolModa, unfortunately, cannot dock in the presence of cofactors (e.g. NADPH), which could impact results of our virtual screening. Thus, the instructor will provide docking results with the NADPH cofactor while you use MolModa to examine the impact of its removal.
Important
We recommend that you use Google Chrome instead of other browsers. Some browsers (e.g., Firefox) have been shown to exhibit more bugs and not work smoothly.
Protein preparation¶
Here are the general steps in MolModa to prepare the protein for docking.
- Download this specific
3FRDPDB structure (click this link!) and import it into MolModa usingFile->Open. - Remove all non-polymer atoms (e.g., water molecules and all co-crystallized ligands) from the protein structure.
- Protonate the protein at a pH of 7.4.
- Add a region with a center of (-9, 34, -4) and dimensions of (26, 25, 27).
Reference image
After this stage, your MolModa session should look like this. Note that the color of your region may be different.
Ligand preparation¶
Here are the general steps in MolModa to prepare the ligands for docking.
- Download the active molecules' SMILES and load them into MolModa by using
File->Openor by drag and drop. - Protonate all compounds at a pH of 7.4 while regenerating coordinates with the "recommended" 3D coordinates generation option.
Reference image
After this stage, your MolModa session should look like this.
Docking¶
Dock the protonated compounds into the 3FRD protein using an exhaustiveness of 24 in MolModa.
These are your docking results for the active compounds without NADPH.
Reference image
After this stage, your MolModa session should look like this.
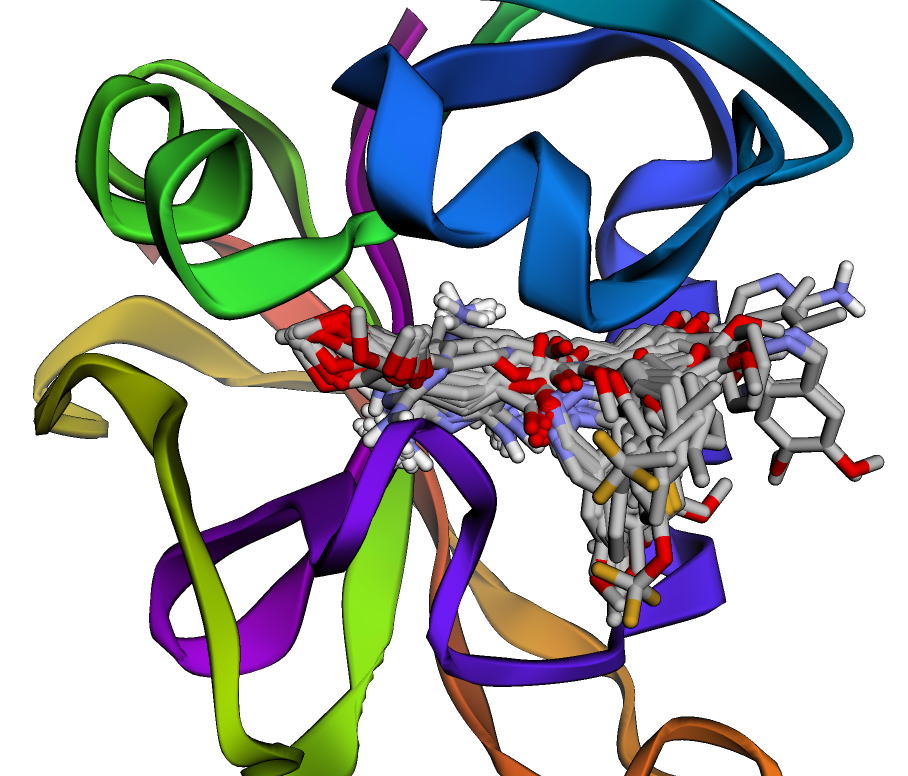
You need to download your docked ligand poses by going to File -> Save, uncheck "Save in MolModa format", and use these settings.
This will download a zip file that you can extract and have all of your docked poses as mol2 files.
You can load these compounds into PyMOL.
Submission
In your submission, answer the following questions:
- Download and extract on your computer these docking results that were performed in the presence of NADPH using AutoDock Vina.
In PyMOL, load the
3frd.pdbstructure and all active.pdbqtfiles in theposes/directory. These.pdbqtfiles contain the top 9 poses of the identified binding modes. To see their scores, look at the respective log file in thelogs/directory. Choose any three active compounds and compare these poses to your results—you can download your ligand poses from MolModa by clickingFile->Saveand unchecking "Save project in .molmoda format". Be sure to look at more than just the top pose from the NADPH results. Use screenshots to justify your observations. (PyMol session file)
Analysis¶
As mentioned in most CSB lectures, the purpose of docking is to be able to identify potentially active compounds out of the massively large chemical space. Below is a table from Muddala et al. that elucidates the binding affinity and antibacterial properties of novel ligands to S. aureus DHFR.
| Label | Inhibitor | Average IC50 ± SEM (nM) | Average Ki ± SEM (nM) | MIC (µg/mL) |
|---|---|---|---|---|
| 11a | CHEMBL2042372 |
6.3 ± 0.3 | 1.2 ± 0.1 | 0.0625-0.25 |
| 11b | CHEMBL2042373 |
5.5 ± 1.0 | 1.1 ± 0.2 | 0.0469-0.0938 |
| 11c | CHEMBL2042374 |
8.7 ± 2.1 | 1.7 ± 0.4 | 0.0938-0.1875 |
| 11d | CHEMBL2042375 |
8.2 ± 0.5 | 1.6 ± 0.1 | 0.0938-0.25 |
| 11e | CHEMBL2042376 |
9.2 ± 0.5 | 1.8 ± 0.1 | 0.125-0.25 |
| 11f | CHEMBL2042377 |
13.1 ± 3.1 | 2.6 ± 0.6 | 0.5 |
| 11g | CHEMBL551038 |
4.6 ± 0.7 | 0.9 ± 0.1 | 0.1875-0.375 |
| 11h | CHEMBL565020 |
4.1 ± 0.4 | 0.8 ± 0.1 | 0.125-0.25 |
| 11i | CHEMBL2042475 |
5.8 ± 0.3 | 1.1 ± 0.1 | 0.125-0.25 |
| 11j | CHEMBL550083 |
8.4 ± 1.7 | 1.7 ± 0.3 | 0.0469-0.1875 |
| 11k | CHEMBL2042476 |
7.2 ± 0.8 | 1.4 ± 0.2 | 0.25 |
| 11l | CHEMBL2042477 |
4.8 ± 0.8 | 0.9 ± 0.2 | 0.125 |
| 11m | CHEMBL2042478 |
5.7 ± 0.6 | 1.1 ± 0.1 | 0.125-0.375 |
| 11n | CHEMBL2042479 |
7.0 ± 0.3 | 1.4 ± 0.1 | 0.25-0.5 |
| 11o | CHEMBL2042480 |
11.6 ± 2.2 | 2.3 ± 0.4 | 0.5 |
| 11p | CHEMBL2042481 |
6.3 ± 1.9 | 1.2 ± 0.4 | 0.5 |
| 11q | CHEMBL2042482 |
9.6 ± 0.5 | 1.9 ± 0.1 | 1 |
| 12a | N/A | 7.7 ± 2.9 | 1.5 ± 0.6 | 1 |
| 12b | CHEMBL4799218 |
5.8 ± 3.3 | 1.1 ± 0.7 | 1 |
| 12c | CHEMBL4781757 |
3.3 ± 1.7 | 0.7 ± 0.3 | 1 |
| 12d | N/A | 6.7 ± 3.9 | 1.3 ± 0.8 | 0.5-1 |
| 12e | N/A | 6.3 ± 2.8 | 1.2 ± 0.6 | 0.5 |
| 12f | N/A | 5.6 ± 2.8 | 1.1 ± 0.6 | 0.5 |
| 12g | N/A | 4.5 ± 2.6 | 0.9 ± 0.5 | 1 |
| 12h | N/A | 4.5 ± 2.2 | 0.9 ± 0.4 | 1 |
| 12i | N/A | 5.4 ± 3.1 | 1.1 ± 0.6 | 0.5 |
| 12j | N/A | 5.2 ± 2.3 | 1.0 ± 0.5 | 0.5 |
| 13a | N/A | 8.4 ± 3.8 | 1.7 ± 0.7 | 0.5-1 |
| 13b | CHEMBL4790501 |
6.1 ± 2.7 | 1.2 ± 0.5 | 0.5-1 |
| 13c | N/A | 5.2 ± 3.0 | 1.0 ± 0.6 | 0.5 |
| 13d | N/A | 7.2 ± 3.2 | 1.4 ± 0.6 | 0.5 |
| 13e | N/A | 7.4 ± 4.2 | 1.5 ± 0.8 | 0.5 |
| 13f | N/A | 6.1 ± 2.7 | 1.2 ± 0.5 | 0.5 |
Your instructor has docked all of these active ligands (in addition to the canonical reactant, DFH) and 100 randomly selected decoys from FDA approved drugs into 3FRD, 6PRA, and 6PR6.
You can download the results here: 3FRD-results.zip, 6PRA-results.zip, 6PR6-results.zip.
Submission
In your submission, answer the following questions:
- What is the meaning/significance of IC50, Ki, and MIC? For an active molecule, what are the ideal values for these properties?
3FRDis a crystal structure with NADPH and DHF, which should be an optimal protein structure to design a DHF competitive inhibitor. Is there a correlation between AutoDock Vina docking scores (with NADPH) and experimental Ki values? Quantify this correlation with an R2 value.6PRAis a crystal structure that does not have DHF. Explain how this could impact docking calculations. Compare the docking scores of active ligands from6PRAand3FRD; are there significant differences? (PyMol session file)6PR6was crystallized with the active compound11j. Compare the crystallized ligand pose to the top 9 poses and scores of the same11jligand. What do you observe? Support your claims with screenshots. (PyMol session file)- Decoy ligands provide a mechanism to ensure that virtual screening workflows work as expected.
Using the
3FRDresults, examine the decoy scores and poses with the top three best and worst scores. Does our pipeline accurately sort these decoy molecules? Why or why not? Examine the top poses of these best and worst decoys. Are there any noncovalent interaction trends? Support your claims with screenshots.
Ended: Project
Ended: Assessments
Team¶
Alex Maldonado (Instructor)¶

Address me as: Alex (preferred), Dr. Maldonado, Dr. Alex, Dr. M.
Pronouns: he/him/his
Major: BSE (Western Michigan University) and PhD in Chemical Engineering (University of Pittsburgh)
Level: Postdoctoral Associate in Computational Biology
Contact: alex.maldonado@pitt.edu for most communication.
I generally respond within 12 hours.
Office hours:
| Day | Time | Location |
|---|---|---|
| Tuesday | 11:30 am - 12:30 pm | 102 Clapp Hall |
| Thursday | 11:30 am - 12:30 pm | 315 Clapp Hall |
Office: 103 Clapp Hall
Ask me about . . . recent films I've watched, my favorite Pittsburgh restaurants, my cat, my Spotify On Repeat.
Research . . . Undergraduate: immunodiagnostics Graduate: quantum chemistry, machine learning, molecular simulations Postdoc: force field parameterization, protein-ligand binding, drug discovery.
Teaching assistants¶
Contact information can be found on Canvas.
Reya Kundu¶
Address me as: Reya
Pronouns: she/her/hers
Major: Computational Biology
Level: Senior
Office hours: Wednesdays 9:00 - 10:00 am in 102 Clapp
Ask me about: language learning (Swedish and Turkish), neurobiology research, horror movies, Bollywood.
Career trajectory: Pre-med
Justine Denby¶
Address me as: Justine
Pronouns: she/her/hers
Major: Computational Biology
Level: Junior
Office hours: Tuesdays 2:00 - 3:00 pm in L01 Clapp
Ask me about Working out, climbing, music, video games, and reading!
Career trajectory PhD and industry
Students¶
Level¶
| Freshman | Sophomore | Junior | Senior |
|---|---|---|---|
| 0 | 13 | 31 | 31 |
Programs of study¶
These include majors and minors of students enrolled in the class.
- Computational Biology: 43
- Chemistry: 33
- Conceptual Fds of Medicine: 13
- Biological Sciences: 10
- Undeclared: 8
- Biochemistry: 6
- Molecular Biology: 5
- Studio Arts: 4
- Economics: 3
- Applied Statistics: 3
- French: 2
- Computer Science: 2
- Neuroscience: 2
- History: 2
- Korean: 2
- Philosophy: 1
- Gender, Sexuality & Women's St: 1
- Japanese: 1
- Public and Professional Writng: 1
- Statistics: 1
- Digital Narr & Interact Design: 1
- Ecology and Evolution: 1
- English Writing: 1
- Industrial Engineering: 1
- Sociology: 1
- Italian: 1
- Children's Literature: 1
- Religious Studies: 1
Programming+ recitations¶
Welcome to the Programming+ recitations for our Computational Biology course!
| Day | Time | Location |
|---|---|---|
| Friday | 2:00 - 3:30 pm | 315 Clapp Hall |
What are Programming+ Problems?¶
Programming+ problems are optional, advanced coding challenges designed to:
- Deepen your understanding of computational biology concepts
- Enhance your programming skills
- Provide hands-on experience with real-world bioinformatics scenarios
These problems are not required for course completion but offer valuable additional learning opportunities.
Recitation Format¶
These sessions are not lectures. Instead, they provide a dedicated time and space for you to:
- Ask questions about Programming+ problems
- Seek guidance on approach and implementation
- Discuss challenges and potential solutions
- Collaborate with other like-minded students
The instructor will be available to provide support, clarification, and direction as needed.
What to Bring¶
- Your laptop
- Any code you're working on
- Specific questions or issues you'd like to address
Guidelines¶
- Come prepared: Review the problem and attempt a solution before the recitation.
- Be specific: The more precise your questions, the more effectively we can assist you.
- Collaborate: Feel free to discuss approaches with your peers during the session.
- Respect others: Keep conversations at a reasonable volume.
We look forward to seeing you at the recitations and supporting your journey in computational biology!